
Cell Reprogramming for Regeneration and Repair of the Nervous System

 , , , , ,
, , , , ,
Abstract
:1. Introduction
1.1. Cell Reprogramming
1.2. Cell Reprogramming to Generate Neurons
2. Genetic Engineering for Cell Reprogramming
2.1. Vector and Promoter Design
2.2. Retroviruses and Lentiviruses
2.3. Adeno-Associated Viruses
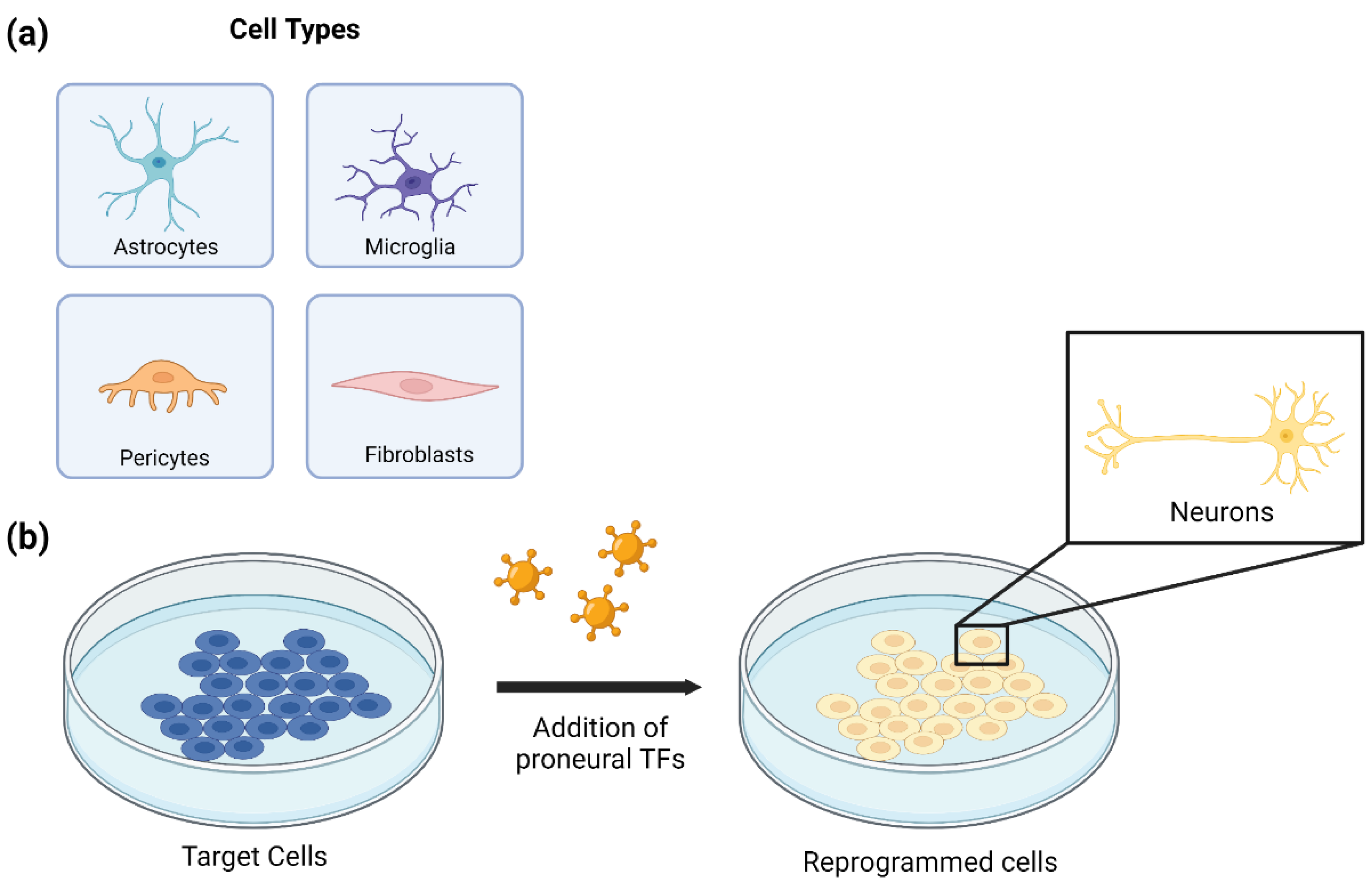
3. In Vitro Cell Reprogramming for Generating Neuronal Cells
3.1. Astrocyte to Neuron Reprogramming
3.2. Microglia to Neuron Reprogramming
3.3. Pericyte to Neuron Reprogramming
3.4. Fibroblast to Neuron Reprogramming
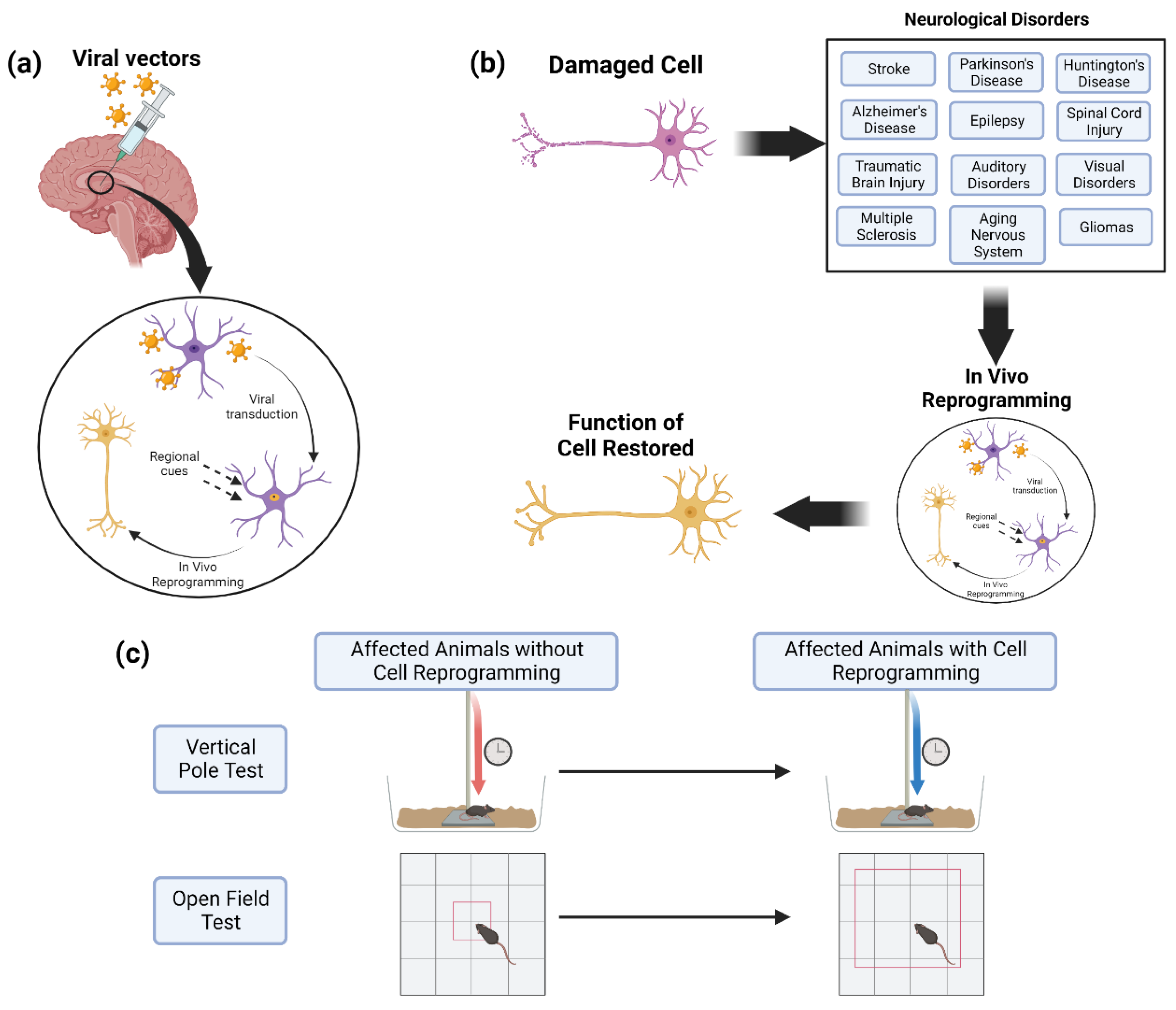
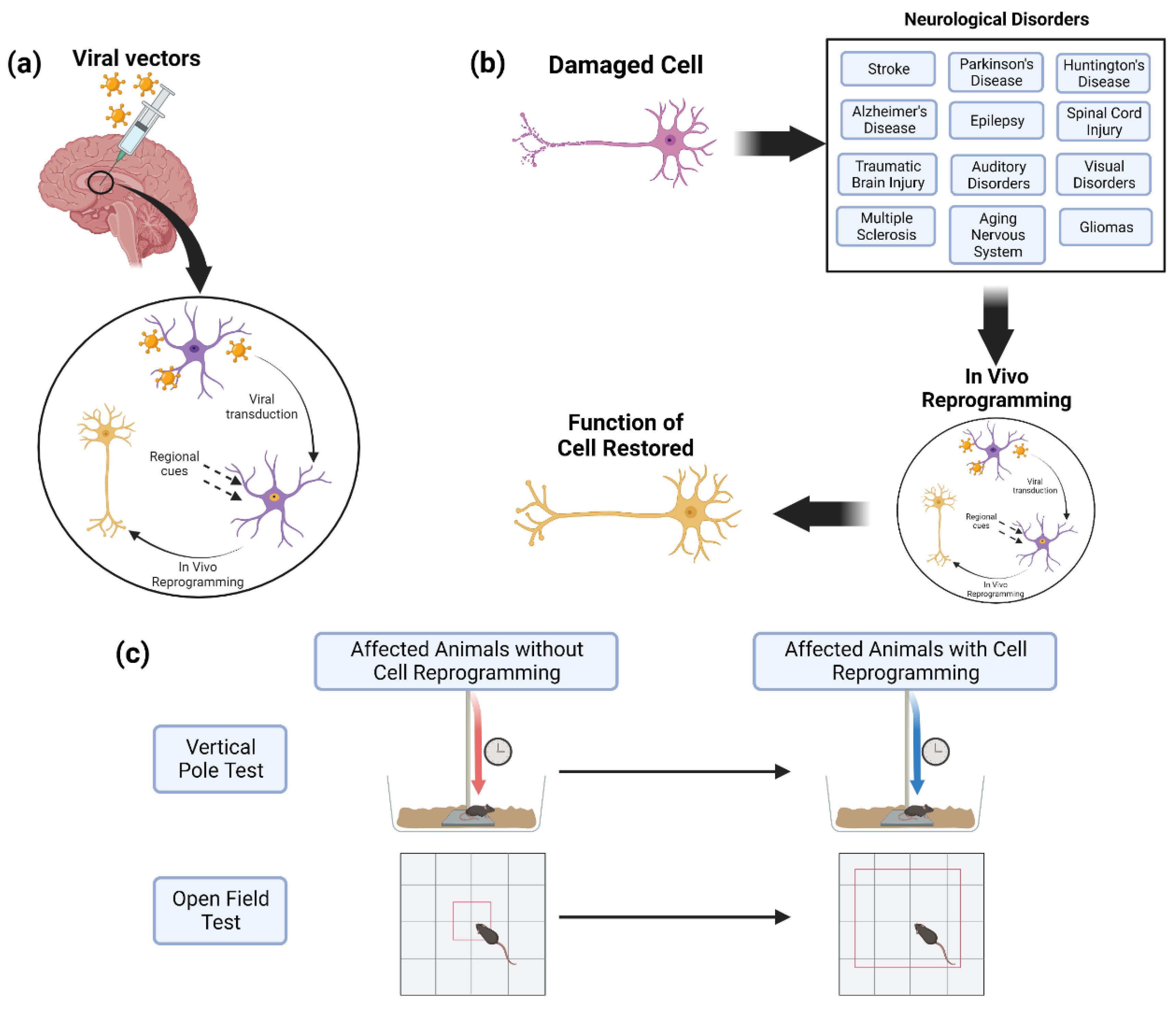
4. In Vivo Cell Reprogramming for Neurological Disorders
4.1. Stroke
4.2. Parkinson’s Disease
4.3. Huntington’s Disease
4.4. Alzheimer’s Disease
4.5. Epilepsy
4.6. Spinal Cord Injury
4.7. Traumatic Brain Injury
4.8. Auditory Disorders
4.9. Visual Disorders
4.10. Multiple Sclerosis
4.11. Aging of the Nervous System
4.12. Gliomas
5. Challenges
5.1. In Vitro Challenges
5.2. In Vivo Reprogramming vs. Neuroprotection
5.3. In Vivo Viability
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascón, S.; Masserdotti, G.; Russo, G.L.; Götz, M. Direct Neuronal Reprogramming: Achievements, Hurdles, and New Roads to Success. Cell Stem Cell 2017, 21, 18–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Cheng, C.; Liu, Y.; Liu, N.; Lo, E.H.; Wang, X. Neuroglobin Promotes Neurogenesis through Wnt Signaling Pathway. Cell Death Dis. 2018, 9, 945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, R.A.; Barrett, J.; Mason, S.L.; Björklund, A. Fetal Dopaminergic Transplantation Trials and the Future of Neural Grafting in Parkinson’s Disease. Lancet Neurol. 2013, 12, 84–91. [Google Scholar] [CrossRef]
- Kefalopoulou, Z.; Politis, M.; Piccini, P.; Mencacci, N.; Bhatia, K.; Jahanshahi, M.; Widner, H.; Rehncrona, S.; Brundin, P.; Björklund, A.; et al. Long-Term Clinical Outcome of Fetal Cell Transplantation for Parkinson Disease Two Case Reports. JAMA Neurol. 2014, 71, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martín-Montañez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.-H.; et al. Induction of Functional Dopamine Neurons from Human Astrocytes in Vitro and Mouse Astrocytes in a Parkinson’s Disease Model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- Kelaini, S.; Cochrane, A.; Margariti, A. Direct Reprogramming of Adult Cells: Avoiding the Pluripotent State. Stem Cells Cloning Adv. Appl. 2014, 7, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, C.; Blum, R.; Gascón, S.; Masserdotti, G.; Tripathi, P.; Sánchez, R.; Tiedt, S.; Schroeder, T.; Götz, M.; Berninger, B. Directing Astroglia from the Cerebral Cortex into Subtype Specific Functional Neurons. PLoS Biol. 2010, 8, e1000373. [Google Scholar] [CrossRef] [Green Version]
- Masserdotti, G.; Gillotin, S.; Sutor, B.; Drechsel, D.; Irmler, M.; Jørgensen, H.F.; Sass, S.; Theis, F.J.; Beckers, J.; Berninger, B.; et al. Transcriptional Mechanisms of Proneural Factors and REST in Regulating Neuronal Reprogramming of Astrocytes. Cell Stem Cell 2015, 17, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Qin, S.; Huang, X.; Yuan, Y.; Tan, Z.; Gu, Y.; Cheng, X.; Wang, D.; Lian, X.-F.; He, C.; et al. Region-Restrict Astrocytes Exhibit Heterogeneous Susceptibility to Neuronal Reprogramming. Stem Cell Rep. 2019, 12, 290–304. [Google Scholar] [CrossRef]
- Berninger, B.; Costa, M.R.; Koch, U.; Schroeder, T.; Sutor, B.; Grothe, B.; Götz, M. Functional Properties of Neurons Derived from In Vitro Reprogrammed Postnatal Astroglia. J. Neurosci. 2007, 27, 8654–8664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchane, M.; Melo de Farias, A.R.; Maria de Sousa Moura, D.; Hilscher, M.M.; Schroeder, T.; Leão, R.N.; Costa, M.R. Lineage Reprogramming of Astroglial Cells from Different Origins into Distinct Neuronal Subtypes. Stem Cell Rep. 2017, 9, 162–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Zhou, W.; Jin, H.; Li, T. Brn2 Alone Is Sufficient to Convert Astrocytes into Neural Progenitors and Neurons. Stem Cells Dev. 2018, 27, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Gascón, S.; Murenu, E.; Masserdotti, G.; Ortega, F.; Russo, G.L.; Petrik, D.; Deshpande, A.; Heinrich, C.; Karow, M.; Robertson, S.P.; et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell 2016, 18, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Zhang, L.; Wu, Z.; Chen, Y.; Wang, F.; Chen, G. In Vivo Direct Reprogramming of Reactive Glial Cells into Functional Neurons after Brain Injury and in an Alzheimer’s Disease Model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar] [CrossRef] [Green Version]
- Brulet, R.; Matsuda, T.; Zhang, L.; Miranda, C.; Giacca, M.; Kaspar, B.K.; Nakashima, K.; Hsieh, J. NEUROD1 Instructs Neuronal Conversion in Non-Reactive Astrocytes. Stem Cell Rep. 2017, 8, 1506–1515. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Ma, N.-X.; Pei, Z.-F.; Wu, Z.; Do-Monte, F.H.; Keefe, S.; Yellin, E.; Chen, M.S.; Yin, J.-C.; Lee, G.; et al. A NeuroD1 AAV-Based Gene Therapy for Functional Brain Repair after Ischemic Injury through In Vivo Astrocyte-to-Neuron Conversion. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wu, Q.; Gao, M.; Ryu, E.; Pei, Z.; Kissinger, S.T.; Chen, Y.; Rao, A.K.; Xiang, Z.; Wang, T. Restoration of Visual Function and Cortical Connectivity After Ischemic Injury Through NeuroD1-Mediated Gene Therapy. Front. Cell Dev. Biol. 2021, 9, 720078. [Google Scholar] [CrossRef]
- Puls, B.; Ding, Y.; Zhang, F.; Pan, M.; Lei, Z.; Pei, Z.; Jiang, M.; Bai, Y.; Forsyth, C.; Metzger, M.; et al. Regeneration of Functional Neurons After Spinal Cord Injury via in situ NeuroD1-Mediated Astrocyte-to-Neuron Conversion. Front. Cell Dev. Biol. 2020, 8, 591883. [Google Scholar] [CrossRef]
- Khanghahi, A.M.; Satarian, L.; Deng, W.; Baharvand, H.; Javan, M. In Vivo Conversion of Astrocytes into Oligodendrocyte Lineage Cells with Transcription Factor Sox10.; Promise for Myelin Repair in Multiple Sclerosis. PLoS ONE 2018, 13, e0203785. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, J.-C.; Yeh, H.; Ma, N.-X.; Lee, G.; Chen, X.A.; Wang, Y.; Lin, L.; Chen, L.; Jin, P.; et al. Small Molecules Efficiently Reprogram Human Astroglial Cells into Functional Neurons. Cell Stem Cell 2015, 17, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.-C.; Zhang, L.; Ma, N.-X.; Wang, Y.; Lee, G.; Hou, X.-Y.; Lei, Z.-F.; Zhang, F.-Y.; Dong, F.-P.; Wu, G.-Y.; et al. Chemical Conversion of Human Fetal Astrocytes into Neurons through Modulation of Multiple Signaling Pathways. Stem Cell Rep. 2019, 12, 488–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Tao, X.; Sui, M.; Cui, M.; Liu, S.; Wang, B.; Wang, T.; Zheng, Y.; Luo, J.; Mu, Y.; et al. Reprogramming astrocytes to motor neurons by activation of endogenous Neurog2 and Isl1. Stem Cell Rep. 2021, 16, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Parry, M.; Hou, X.-Y.; Liu, M.-H.; Wang, H.; Cain, R.; Pei, Z.-F.; Chen, Y.-C.; Gui, Z.-Y.; Abhijeet, S.; et al. Gene Therapy Conversion of Striatal Astrocytes into GABAergic Neurons in Mouse Models of Huntington’s Disease. Nat. Commun. 2020, 11, 1105. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Shang, J.; Nakano, Y.; Morihara, R.; Sato, K.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; Abe, K. In Vivo Direct Reprogramming of Glial Linage to Mature Neurons after Cerebral Ischemia. Sci. Rep. 2019, 9, 10956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresita, A.; Glavan, D.; Udristoiu, I.; Catalin, B.; Hermann, D.M.; Popa-Wagner, A. Very Low Efficiency of Direct Reprogramming of Astrocytes Into Neurons in the Brains of Young and Aged Mice After Cerebral Ischemia. Front. Aging Neurosci. 2019, 11, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentini, C.; d’Orange, M.; Marichal, N.; Trottmann, M.-M.; Vignoles, R.; Foucault, L.; Verrier, C.; Massera, C.; Raineteau, O.; Conzelmann, K.-K.; et al. Reprogramming Reactive Glia into Interneurons Reduces Chronic Seizure Activity in a Mouse Model of Mesial Temporal Lobe Epilepsy. Cell Stem Cell 2021, 28, 2104–2121.e10. [Google Scholar] [CrossRef]
- Mattugini, N.; Bocchi, R.; Scheuss, V.; Russo, G.L.; Torper, O.; Lao, C.L.; Götz, M. Inducing Different Neuronal Subtypes from Astrocytes in the Injured Mouse Cerebral Cortex. Neuron 2019, 103, 1086–1095.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020, 181, 590–603.e16. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a Model of Parkinson’s Disease with in Situ Converted Nigral Neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Ghasemi-Kasman, M.; Shojaei, A.; Gol, M.; Moghadamnia, A.A.; Baharvand, H.; Javan, M. MiR-302/367-Induced Neurons Reduce Behavioral Impairment in an Experimental Model of Alzheimer’s Disease. Mol. Cell. Neurosci. 2018, 86, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Irie, T.; Katsurabayashi, S.; Hayashi, Y.; Nagai, T.; Hamazaki, N.; Adefuin, A.M.D.; Miura, F.; Ito, T.; Kimura, H.; et al. Pioneer Factor NeuroD1 Rearranges Transcriptional and Epigenetic Profiles to Execute Microglia-Neuron Conversion. Neuron 2019, 101, 472–485.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Wang, X.; Xiong, W.; Chen, J. In Vivo Reprogramming Reactive Glia into IPSCs to Produce New Neurons in the Cortex Following Traumatic Brain Injury. Sci. Rep. 2016, 6, 22490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karow, M.; Sánchez, R.; Schichor, C.; Masserdotti, G.; Ortega, F.; Heinrich, C.; Gascón, S.; Khan, M.A.; Lie, D.C.; Dellavalle, A.; et al. Reprogramming of Pericyte-Derived Cells of the Adult Human Brain into Induced Neuronal Cells. Cell Stem Cell 2012, 11, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Karow, M.; Schichor, C.; Beckervordersandforth, R.; Berninger, B. Lineage-Reprogramming of Pericyte-Derived Cells of the Adult Human Brain into Induced Neurons. J. Vis. Exp. 2014, 87, e51433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karow, M.; Camp, J.G.; Falk, S.; Gerber, T.; Pataskar, A.; Gac-Santel, M.; Kageyama, J.; Brazovskaja, A.; Garding, A.; Fan, W.; et al. Direct Pericyte-to-Neuron Reprogramming via Unfolding of a Neural Stem Cell-like Program. Nat. Neurosci. 2018, 21, 932–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, P.; Zhang, S.; Yeap, Y.; Feng, Z. Induction of neuronal phenotypes from NG2+ glial progenitors by inhibiting epidermal growth factor receptor in mouse spinal cord injury. Glia 2012, 60, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Björklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct Conversion of Human Fibroblasts to Dopaminergic Neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct Generation of Functional Dopaminergic Neurons from Mouse and Human Fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef]
- Hu, W.; Qiu, B.; Guan, W.; Wang, Q.; Wang, M.; Li, W.; Gao, L.; Shen, L.; Huang, Y.; Xie, G.; et al. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell 2015, 17, 204–212. [Google Scholar] [CrossRef]
- Arnold, A.; Naaldijk, Y.M.; Fabian, C.; Wirth, H.; Binder, H.; Nikkhah, G.; Armstrong, L.; Stolzing, A. Reprogramming of Human Huntington Fibroblasts Using MRNA. ISRN Cell Biol. 2011, 2012, e124878. [Google Scholar] [CrossRef] [Green Version]
- García-León, J.A.; Kumar, M.; Boon, R.; Chau, D.; One, J.; Wolfs, E.; Eggermont, K.; Berckmans, P.; Gunhanlar, N.; de Vrij, F.; et al. SOX10 Single Transcription Factor-Based Fast and Efficient Generation of Oligodendrocytes from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 655–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.-H.; Jeong, Y.-W.; Choi, W.; Noh, J.-E.; Lee, S.; Kim, H.-S.; Song, J. Multimodal Therapeutic Effects of Neural Precursor Cells Derived from Human-Induced Pluripotent Stem Cells through Episomal Plasmid-Based Reprogramming in a Rodent Model of Ischemic Stroke. Stem Cells Int. 2020, 2020, 4061516. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.X.; Yuan, Q.; Tan, S.; Xiao, Y.; Wang, D.; Khoo, A.T.T.; Sani, L.; Tran, H.-D.; Kim, P.; Chiew, Y.S.; et al. Direct Induction and Functional Maturation of Forebrain GABAergic Neurons from Human Pluripotent Stem Cells. Cell Rep. 2016, 16, 1942–1953. [Google Scholar] [CrossRef] [Green Version]
- Matjusaitis, M.; Wagstaff, L.J.; Martella, A.; Baranowski, B.; Blin, C.; Gogolok, S.; Williams, A.; Pollard, S.M. Reprogramming of Fibroblasts to Oligodendrocyte Progenitor-like Cells Using CRISPR/Cas9-Based Synthetic Transcription Factors. Stem Cell Rep. 2019, 13, 1053–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Weichert, R.M.; Liu, W.; Davis, R.L.; Dabdoub, A. Generation of Induced Neurons by Direct Reprogramming in the Mammalian Cochlea. Neuroscience 2014, 275, 125–135. [Google Scholar] [CrossRef]
- Cheng, X.; Tan, Z.; Huang, X.; Yuan, Y.; Qin, S.; Gu, Y.; Wang, D.; He, C.; Su, Z. Inhibition of Glioma Development by ASCL1-Mediated Direct Neuronal Reprogramming. Cells 2019, 8, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; He, H.; Zhou, K.; Ren, Y.; Shi, Z.; Wu, Z.; Wang, Y.; Lu, Y.; Jiao, J. Neuronal Transcription Factors Induce Conversion of Human Glioma Cells to Neurons and Inhibit Tumorigenesis. PLoS ONE 2012, 7, e41506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Zang, T.; Liu, M.-L.; Wang, L.-L.; Niu, W.; Zhang, C.-L. Reprogramming the Fate of Human Glioma Cells to Impede Brain Tumor Development. Cell Death Dis. 2014, 5, e1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and Mutation Independent Therapy via CRISPR-Cas9 Mediated Cellular Reprogramming in Rod Photoreceptors. Cell Res. 2017, 27, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Masserdotti, G.; Gascón, S.; Götz, M. Direct Neuronal Reprogramming: Learning from and for Development. Development 2016, 143, 2494–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouaux, C.; Arlotta, P. Direct Lineage Reprogramming of Post-Mitotic Callosal Neurons into Corticofugal Neurons In Vivo. Nat. Cell Biol. 2013, 15, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.F.; Terry, A.; Beretta, C.; Pereira, C.F.; Leleu, M.; Chen, Z.-F.; Kelly, C.; Merkenschlager, M.; Fisher, A.G. REST Selectively Represses a Subset of RE1-Containing Neuronal Genes in Mouse Embryonic Stem Cells. Development 2009, 136, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossen, M.; Bujard, H. Tight Control of Gene Expression in Mammalian Cells by Tetracycline-Responsive Promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colasante, G.; Lignani, G.; Rubio, A.; Medrihan, L.; Yekhlef, L.; Sessa, A.; Massimino, L.; Giannelli, S.G.; Sacchetti, S.; Caiazzo, M.; et al. Rapid Conversion of Fibroblasts into Functional Forebrain GABAergic Interneurons by Direct Genetic Reprogramming. Cell Stem Cell 2015, 17, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Südhof, T.C.; Wernig, M. Direct Conversion of Fibroblasts to Functional Neurons by Defined Factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Lakso, M.; Sauer, B.; Mosinger, B., Jr.; Lee, E.J.; Manning, R.W.; Yu, S.H.; Mulder, K.L.; Westphal, H. Targeted Oncogene Activation by Site-Specific Recombination in Transgenic Mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6232–6236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orban, P.C.; Chui, D.; Marth, J.D. Tissue- and Site-Specific DNA Recombination in Transgenic Mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6861–6865. [Google Scholar] [CrossRef] [Green Version]
- Sauer, B.; Henderson, N. Site-Specific DNA Recombination in Mammalian Cells by the Cre Recombinase of Bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandoski, M. Conditional Control of Gene Expression in the Mouse. Nat. Rev. Genet. 2001, 2, 743–755. [Google Scholar] [CrossRef]
- Nagy, A. Cre Recombinase: The Universal Reagent for Genome Tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR Interference (CRISPRi) for Sequence-Specific Control of Gene Expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, L.F.; Vanderhaeghen, J.J.; Bignami, A.; Gerstl, B. An Acidic Protein Isolated from Fibrous Astrocytes. Brain Res. 1971, 28, 351–354. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Barriers to Neurotoxic Inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Cañedo, A.; Bonamino, M.H. Retroviral Vectors and Transposons for Stable Gene Therapy: Advances, Current Challenges and Perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral Vectors: Basic to Translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Hindmarsh, P.; Leis, J. Retroviral DNA Integration. Microbiol. Mol. Biol. Rev. MMBR 1999, 63, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Merienne, N.; Le Douce, J.; Faivre, E.; Déglon, N.; Bonvento, G. Efficient Gene Delivery and Selective Transduction of Astrocytes in the Mammalian Brain Using Viral Vectors. Front. Cell. Neurosci. 2013, 7, 106. [Google Scholar] [CrossRef]
- Colin, A.; Faideau, M.; Dufour, N.; Auregan, G.; Hassig, R.; Andrieu, T.; Brouillet, E.; Hantraye, P.; Bonvento, G.; Déglon, N. Engineered Lentiviral Vector Targeting Astrocytes In Vivo. Glia 2009, 57, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Miletic, H.; Fischer, Y.H.; Neumann, H.; Hans, V.; Stenzel, W.; Giroglou, T.; Hermann, M.; Deckert, M.; Von Laer, D. Selective Transduction of Malignant Glioma by Lentiviral Vectors Pseudotyped with Lymphocytic Choriomeningitis Virus Glycoproteins. Hum. Gene Ther. 2004, 15, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous MicroRNA Can Be Broadly Exploited to Regulate Transgene Expression According to Tissue, Lineage and Differentiation State. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of Small DNA Viruses Found in Various Adenovirus Preparations: Physical, Biological, and Immunological Characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-Specific Integration by Adeno-Associated Virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [Green Version]
- Samulski, R.J.; Zhu, X.; Xiao, X.; Brook, J.D.; Housman, D.E.; Epstein, N.; Hunter, L.A. Targeted Integration of Adeno-Associated Virus (AAV) into Human Chromosome 19. EMBO J. 1991, 10, 3941–3950. [Google Scholar] [CrossRef]
- Hermonat, P.L. The First Adeno-Associated Virus Gene Transfer Experiment, 1983. Hum. Gene Ther. 2014, 25, 486–487. [Google Scholar] [CrossRef]
- Xiao, X.; Li, J.; Samulski, R.J. Efficient Long-Term Gene Transfer into Muscle Tissue of Immunocompetent Mice by Adeno-Associated Virus Vector. J. Virol. 1996, 70, 8098–8108. [Google Scholar] [CrossRef] [Green Version]
- FDA. FDA Approves Novel Gene Therapy to Treat Patients with a Rare Form of Inherited Vision Loss.; FDA: Silver Spring, MD, USA, 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (accessed on 7 September 2020).
- Gao, G.; Vandenberghe, L.H.; Wilson, J.M. New Recombinant Serotypes of AAV Vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 Preferentially Targets Neonatal Neurons and Adult Astrocytes. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkel, S.F.; Andrews, A.M.; Lutton, E.M.; Mu, D.; Hudry, E.; Hyman, B.T.; Maguire, C.A.; Ramirez, S.H. Trafficking of Adeno-Associated Virus Vectors across a Model of the Blood–Brain Barrier.; a Comparative Study of Transcytosis and Transduction Using Primary Human Brain Endothelial Cells. J. Neurochem. 2017, 140, 216–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yang, B.; Mu, X.; Ahmed, S.S.; Su, Q.; He, R.; Wang, H.; Mueller, C.; Sena-Esteves, M.; Brown, R.; et al. Several RAAV Vectors Efficiently Cross the Blood–Brain Barrier and Transduce Neurons and Astrocytes in the Neonatal Mouse Central Nervous System. Mol. Ther. 2011, 19, 1440–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Kumar, S.R.; Adams, C.D.; Yang, D.; Wang, T.; Wolfe, D.A.; Arokiaaraj, C.M.; Ngo, V.; Campos, L.J.; Griffiths, J.A.; et al. Engineered AAVs for Non-Invasive Gene Delivery to Rodent and Non-Human Primate Nervous Systems. Neuron 2022, 110, 2242–2257.e6. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for Efficient Noninvasive Gene Delivery to the Central and Peripheral Nervous Systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, Neurons, Synapses: A Tripartite View on Cortical Circuit Development. Neural Dev. 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Ma, N.-X.; Yin, J.-C.; Chen, G. Transcriptome Analysis of Small Molecule-Mediated Astrocyte-to-Neuron Reprogramming. Front. Cell Dev. Biol. 2019, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Var, S.R.; Byrd-Jacobs, C.A. Role of Macrophages and Microglia in Zebrafish Regeneration. Int. J. Mol. Sci. 2020, 21, 4768. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Strekalova, T.; Ponomarev, E.D. The Role of Neuronal Factors in the Epigenetic Reprogramming of Microglia in the Normal and Diseased Central Nervous System. Front. Cell. Neurosci. 2019, 13, 453. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.-X.; Wu, T.; et al. Repopulated Microglia Are Solely Derived from the Proliferation of Residual Microglia after Acute Depletion. Nat. Neurosci. 2018, 21, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Du, S.; Yang, B.; Wang, Y.; Li, Y.; Li, R.; Zhou, T.; Du, X.; He, Y.; Wang, Y.; et al. NeuroD1 Induces Microglial Apoptosis and Cannot Induce Microglia-to-Neuron Cross-Lineage Reprogramming. Neuron 2021, 109, 4094–4108.e5. [Google Scholar] [CrossRef]
- Pallone, T.L.; Silldorff, E. Pericyte Regulation of Renal Medullary Blood Flow. Exp. Nephrol. 2001, 9, 165–170. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.-M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The Translational Significance of the Neurovascular Unit. J. Biol. Chem. 2017, 292, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.-P.; Rowley, J.E.; Redpath, A.N.; Tilman, J.D.; Fellous, T.G.; Johnson, J.R. Pericytes, Mesenchymal Stem Cells and Their Contributions to Tissue Repair. Pharmacol. Ther. 2015, 151, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birbrair, A.; Zhang, T.; Wang, Z.-M.; Messi, M.L.; Mintz, A.; Delbono, O. Pericytes at the Intersection between Tissue Regeneration and Pathology. Clin. Sci. 2015, 128, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.D.; Watt, F.M. Fibroblast Heterogeneity: Implications for Human Disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Sim, H.; Ahn, H.; Ha, J.; Baek, A.; Jeon, Y.-J.; Son, M.-Y.; Kim, J. Direct Reprogramming to Human Induced Neuronal Progenitors from Fibroblasts of Familial and Sporadic Parkinson’s Disease Patients. Int. J. Stem Cells 2019, 12, 474–483. [Google Scholar] [CrossRef]
- Patel, R.A.G.; White, C.J. Stroke Treatment and Prevention. Prog. Cardiovasc. Dis. 2017, 59, 525–526. [Google Scholar] [CrossRef]
- Catanese, L.; Tarsia, J.; Fisher, M. Acute Ischemic Stroke Therapy Overview. Circ. Res. 2017, 120, 541–558. [Google Scholar] [CrossRef]
- Duris, K.; Splichal, Z.; Jurajda, M. The Role of Inflammatory Response in Stroke Associated Programmed Cell Death. Curr. Neuropharmacol. 2018, 16, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Hassler, R. Zur. Pathologie der Paralysis agitans und des postenzephalitischen Parkinsonismus. J. Psychol. Neurol. 1938, 48, 387–476. [Google Scholar]
- Hornykiewicz, O. The tropical localization and content of noradrenalin and dopamine (3-hydroxytyramine) in the substantia nigra of normal persons and patients with Parkinson’s disease. Wien. Klin. Wochenschr. 1963, 75, 309–312. [Google Scholar] [PubMed]
- Bertler, A.; Rosengren, E. Occurrence and Distribution of Dopamine in Brain and Other Tissues. Experientia 1959, 15, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Ehringer, H.; Hornykiewicz, O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin. Wochenschr. 1960, 38, 1236–1239. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic Risk Factors in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef]
- Chen, H.; Ritz, B. The Search for Environmental Causes of Parkinson’s Disease: Moving Forward. J. Park. Dis. 2018, 8, S9–S17. [Google Scholar] [CrossRef] [Green Version]
- Barbeau, A.; Murphy, G.F.; Sourkes, T.L. Excretion of Dopamine in Diseases of Basal Ganglia. Science 1961, 133, 1706–1707. [Google Scholar] [CrossRef] [PubMed]
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien. Klin. Wochenschr. 1961, 73, 787–788. [Google Scholar]
- Fabbrini, G.; Brotchie, J.M.; Grandas, F.; Nomoto, M.; Goetz, C.G. Levodopa-Induced Dyskinesias. Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-Based Therapies for Parkinson Disease—Past Insights and Future Potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef]
- Dell’Anno, M.T.; Caiazzo, M.; Leo, D.; Dvoretskova, E.; Medrihan, L.; Colasante, G.; Giannelli, S.; Theka, I.; Russo, G.; Mus, L.; et al. Remote Control of Induced Dopaminergic Neurons in Parkinsonian Rats. J. Clin. Investig. 2014, 124, 3215–3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, W.; Zang, T.; Wang, L.-L.; Zou, Y.; Zhang, C.-L. Phenotypic Reprogramming of Striatal Neurons into Dopaminergic Neuron-like Cells in the Adult Mouse Brain. Stem Cell Rep. 2018, 11, 1156–1170. [Google Scholar] [CrossRef] [Green Version]
- Hallett, P.J.; Deleidi, M.; Astradsson, A.; Smith, G.A.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.-L.; et al. Successful Function of Autologous IPSC-Derived Dopamine Neurons Following Transplantation in a Non-Human Primate Model of Parkinson’s Disease. Cell Stem Cell 2015, 16, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human IPS Cell-Derived Dopaminergic Neurons Function in a Primate Parkinson’s Disease Model. Nature 2017, 548, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, V.; Singh, K.; Kumar, S.; Kim, Y.-S.; Lee, Y.-M.; Kim, J.-J. Therapeutic Advances for Huntington’s Disease. Brain Sci. 2020, 10, 43. [Google Scholar] [CrossRef] [Green Version]
- Victor, M.B.; Richner, M.; Olsen, H.; Lee, S.W.; Monteys, A.M.; Ma, C.; Huh, C.J.; Zhang, B.; Davidson, B.L.; Yang, X.W.; et al. Striatal Neurons Directly Converted from Huntington’s Disease Patient Fibroblasts Recapitulate Age-Associated Disease Phenotypes. Nat. Neurosci. 2018, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic Correction of Huntington’s Disease Phenotypes in Induced Pluripotent Stem Cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Weller, J.; Andrew, B. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef]
- Silva, M.V.F.; Loures, C.D.M.G.; Alves, L.C.V.; Cruz de Souza, L.; Borges, K.B.G.; Carvalho, M.D.G. Alzheimer’s Disease: Risk Factors and Potentially Protective Measures. J. Biomed. Sci. 2019, 26, 33. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Herdy, J.R.; Traxler, L.; Schafer, S.T.; Schlachetzki, J.C.M.; Böhnke, L.; Reid, D.A.; Lee, H.; Zangwill, D.; Fernandes, D.P.; et al. Age-Dependent Instability of Mature Neuronal Fate in Induced Neurons from Alzheimer’s Patients. Cell Stem Cell 2021, 28, 1533–1548.e6. [Google Scholar] [CrossRef]
- Amini, E.; Rezaei, M.; Ibrahim, N.M.; Golpich, M.; Ghasemi, R.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ahmadiani, A. A Molecular Approach to Epilepsy Management: From Current Therapeutic Methods to Preconditioning Efforts. Mol. Neurobiol. 2015, 52, 492–513. [Google Scholar] [CrossRef]
- Dimitrijevic, M.R.; Kakulas, B.A. Spinal Cord Injuries, Human Neuropathology and Neurophysiology. Acta Myol. Myopathies Cardiomyopathies Off. J. Mediterr. Soc. Myol. 2020, 39, 353–358. [Google Scholar] [CrossRef]
- Losey, P.; Anthony, D.C. Impact of Vasculature Damage on the Outcome of Spinal Cord Injury: A Novel Collagenase-Induced Model Give New Insights into the Mechanisms Involved. Neural Regen. Res. 2014, 9, 1783–1786. [Google Scholar] [CrossRef]
- Shi, Z.; Yuan, S.; Shi, L.; Li, J.; Ning, G.; Kong, X.; Feng, S. Programmed cell death in spinal cord injury pathogenesis and therapy. Cell Prolif. 2021, 53, e12992. [Google Scholar] [CrossRef]
- Sutherland, T.C.; Mathews, K.J.; Mao, Y.; Nguyen, T.; Gorrie, C.A. Differences in the Cellular Response to Acute Spinal Cord Injury between Developing and Mature Rats Highlights the Potential Significance of the Inflammatory Response. Front. Cell. Neurosci. 2017, 10, 310. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.; Stern, C.; Freund, P.; Schubert, M.; Sutter, R. Wallerian degeneration in cervical spinal cord tracts is commonly seen in routine T2-weighted MRI after traumatic spinal cord injury and is associated with impairment in a retrospective study. Eur. Radiol. 2020, 31, 2923–2932. [Google Scholar] [CrossRef]
- Dyck, S.M.; Soheila, K.-A. Role of chondroitin sulfate proteoglycan signaling in regulating neuroinflammation following spinal cord injury. Neural Regen. Res. 2018, 13, 2080–2082. [Google Scholar] [CrossRef]
- Mohammed, R.; Opara, K.; Lall, R.; Ojha, U.; Xiang, J. Evaluating the effectiveness of anti-Nogo treatment in spinal cord injuries. Neural Dev. 2020, 15, 1. [Google Scholar] [CrossRef]
- Bellák, T.; Fekécs, Z.; Török, D.; Táncos, Z.; Nemes, C.; Tézsla, Z.; Gál, L.; Polgári, S.; Kobolák, J.; Dinnyás, A.; et al. Grafted human induced pluripotent stem cells improve the outcome of spinal cord injury: Modulation of the lesion microenvironment. Sci. Rep. 2020, 10, 22414. [Google Scholar] [CrossRef]
- Pereira, I.M.; Marote, A.; Salgado, A.J.; Silva, N.A. Filling the Gap: Neural Stem Cells as A Promising Therapy for Spinal Cord Injury. Pharmaceuticals 2019, 12, 65. [Google Scholar] [CrossRef] [Green Version]
- Forgione, N.; Fehlings, M.G. Rho-ROCK Inhibition in the Treatment of Spinal Cord Injury. World Neurosurg. 2014, 82, e535–e539. [Google Scholar] [CrossRef]
- Lu, K.; Cho, C.-L.; Liang, C.-L.; Chen, S.-D.; Liliang, P.-C.; Wang, S.-Y.; Chen, H.-J. Inhibition of the MEK/ERK pathway reduces microglial activation and interleukin-1-beta expression in spinal cord ischemia/reperfusion injury in rats. J. Thorac. Cardiovasc. Surg. 2007, 133, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Joung, D.; Truong, V.; Neitzke, C.C.; Guo, S.-Z.; Walsh, P.J.; Monat, J.R.; Meng, F.; Park, S.H.; Dutton, J.R.; Parr, A.M.; et al. 3D Printed Stem-Cell Derived Neural Progenitors Generate Spinal Cord Scaffolds. Adv. Funct. Mater. 2020, 28, 1801850. [Google Scholar] [CrossRef]
- Tai, W.; Wu, W.; Wang, L.-L.; Ni, H.; Chen, C.; Yang, J.; Zang, T.; Zou, Y.; Xu, X.-M.; Zhang, C.-L. In vivo reprogramming of NG2 glia enables adult neurogenesis and functional recovery following spinal cord injury. Cell Stem Cell 2021, 28, 923–937. [Google Scholar] [CrossRef]
- Wang, L.-L.; Serrano, C.; Zhong, X.; Ma, S.; Zou, Y.; Zhang, C.-L. Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell 2021, 184, 5465–5481. [Google Scholar] [CrossRef]
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.-R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, C.; Bergami, M.; Gascón, S.; Lepier, A.; Viganò, F.; Dimou, L.; Sutor, B.; Berninger, B.; Götz, M. Sox2Sox2-Mediated Conversion of NG2 Glia into Induced Neurons in the Injured Adult Cerebral Cortex. Stem Cell Rep. 2014, 3, 1000–1014. [Google Scholar] [CrossRef] [Green Version]
- Kempfle, J.S.; Luu, N.-N.C.; Petrillo, M.; Al-Asad, R.; Zhang, A.; Edge, A.S.B. Lin28 Reprograms Inner Ear Glia to a Neuronal Fate. Stem Cells 2020, 38, 890–903. [Google Scholar] [CrossRef]
- Noda, T.; Meas, S.J.; Nogami, J.; Amemiya, Y.; Uchi, R.; Ohkawa, Y.; Nishimura, K.; Dabdoub, A. Direct Reprogramming of Spiral Ganglion Non-Neuronal Cells into Neurons: Toward Ameliorating Sensorineural Hearing Loss by Gene Therapy. Front. Cell Dev. Biol. 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Qing, J.; Dong, Y.; Nie, J.; Li, J.; Wang, C.; Lie, Y.; Peng, T.; Duan, M.; Liu, X.; et al. The Role of Transcription Factors of Neurosensory Cells in Non-Syndromic Sensorineural Hearing Loss with or without Inner Ear Malformation. Acta Oto-Laryngol. 2016, 136, 277–282. [Google Scholar] [CrossRef]
- Costa, A.; Sanchez-Guardado, L.; Juniat, S.; Gale, J.E.; Daudet, N.; Henrique, D. Generation of Sensory Hair Cells by Genetic Programming with a Combination of Transcription Factors. Development 2015, 142, 1948–1959. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, A.L.; Rojas-Roldan, L.; Coffin, J. Vision Loss in Older Adults. Am. Fam. Physician 2016, 94, 219–226. [Google Scholar]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the Retinal Vascular Endothelial Cell in Ocular Disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, P.A.; Tang, S.; Shimchuk, A.A.; Ding, S.; Reh, T.A. Potential of Small Molecule–Mediated Reprogramming of Rod Photoreceptors to Treat Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6407–6415. [Google Scholar] [CrossRef] [Green Version]
- Hart, F.M.; Bainbridge, J. Current and Emerging Treatment of Multiple Sclerosis. Am. J. Manag. Care 2016, 22 (Suppl. S6), s159–s170. [Google Scholar]
- Stolt, C.C.; Rehberg, S.; Ader, M.; Lommes, P.; Riethmacher, D.; Schachner, M.; Bartsch, U.; Wegner, M. Terminal Differentiation of Myelin-Forming Oligodendrocytes Depends on the Transcription Factor Sox10. Genes Dev. 2002, 16, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Tamura, N.; Shimazu, K. Aging of the autonomic nervous system. Nihon Rinsho. Jpn. J. Clin. Med. 2005, 63, 973–977. [Google Scholar]
- Segel, M.; Neumann, B.; Hill, M.F.E.; Weber, I.P.; Viscomi, C.; Zhao, C.; Young, A.; Agley, C.C.; Thompson, A.J.; Gonzalez, G.A. Niche Stiffness Underlies the Ageing of Central Nervous System Progenitor Cells. Nature 2019, 573, 130–134. [Google Scholar] [CrossRef]
- Bush, N.A.O.; Chang, S.M.; Berger, M.S. Current and Future Strategies for Treatment of Glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The Epidemiology of Glioma in Adults: A ‘State of the Science’ Review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.-H.; Wu, X.; Tan, F.; Shi, Y.-X.; Glass, T.; Liu, T.J.; Wathen, K.; Hess, K.R.; Gumin, J.; Lang, F.; et al. PAX6 Suppresses Growth of Human Glioblastoma Cells. J. Neuro-Oncol. 2005, 71, 223–229. [Google Scholar] [CrossRef]
- Li, D.-M.; Sun, H. PTEN/MMAC1/TEP1 Suppresses the Tumorigenicity and Induces G1 Cell Cycle Arrest in Human Glioblastoma Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15406–15411. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Manzano, C.; Fueyo, J.; Kyritsis, A.P.; Steck, P.A.; Roth, J.A.; McDonnell, T.J.; Steck, K.D.; Levin, V.A.; Yung, W.K.A. Adenovirus-Mediated Transfer of the P53 Gene Produces Rapid and Generalized Death of Human Glioma Cells via Apoptosis. Cancer Res. 1996, 56, 694–699. [Google Scholar]
- Xu, L.; Xiang, Z.-Q.; Guo, Y.W.; Xu, Y.-G.; Liu, M.-H.; Ji, W.-Y.; He, S.; Lei, W.-L.; Li, W.; Wu, Z.; et al. Enhancing NeuroD1 Expression to Convert Lineage-Traced Astrocytes into Neurons. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lai, M.; Pan, M.; Ge, L.; Liu, J.; Deng, J.; Wang, X.; Li, L.; Wen, J.; Tan, D.; Zhang, H.; et al. NeuroD1 Overexpression in Spinal Neurons Accelerates Axonal Regeneration after Sciatic Nerve Injury. Exp. Neurol. 2020, 327, 113215. [Google Scholar] [CrossRef]
- Zhang, L.; Lei, Z.; Guo, Z.; Pei, Z.; Chen, Y.; Zhang, F.; Cai, A.; Mok, G.; Lee, G.; Swaminathan, V.; et al. Development of Neuroregenerative Gene Therapy to Reverse Glial Scar Tissue Back to Neuron-Enriched Tissue. Front. Cell. Neurosci. 2020, 14, 594170. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Summary Table of Reprogramming Models and Methods | |||
|---|---|---|---|
| Cell Type | Factor(s) | Model | Transduction Details |
| Astrocyte | Neurog2 | In vitro | Retroviral-mediated reprogramming [8] enhanced by REST deletion [9] Lentiviral-mediated reprogramming [10,11] Plasmid transfection-mediated reprogramming [12] |
| Dlx2 | In vitro | Retroviral-mediated reprogramming [8] | |
| Brn2 | In vitro | Retroviral-mediated reprogramming [13] | |
| Ascl1 | In vitro | Lentiviral-mediated reprogramming [11] Plasmid transfection-mediated reprogramming [12] Retroviral-mediated reprogramming enhanced with Bcl-2 expression [14] | |
| NeuroD1 | In vitro Alzheimer’s disease: mouse Ischemic stroke: mouse Spinal cord injury: mouse, rat Non-injured brain: mouse | Retroviral-mediated reprogramming [15] AAV-mediated reprogramming [16] Retroviral-mediated reprogramming [15] AAV-mediated reprogramming [17,18] AAV-mediated reprogramming [19] AAV-mediated reprogramming [16] | |
| Sox10 | Multiple sclerosis demyelination: mouse | Plasmid transfection-mediated, Sox10-based reprogramming [20] | |
| Small Molecule and Combinatorial Approaches | In vitro Huntington’s disease: mouse Ischemic stroke: mouse Epilepsy: mouse Traumatic brain injury: mouse Non-injured spinal cord: mouse | Small molecule-mediated reprogramming with nine-molecule cocktail [21] and four-molecule cocktail [22] Combinatorial Ngn2 and Isl1 CRISPRa-mediated reprogramming [23] Combinatorial NeuroD1 and Dlx2 AAV-mediated reprogramming [24] Combinatorial Ascl1, Sox2, NeuroD1 retroviral-mediated reprogramming [25] Combinatorial Ngn2 and Bcl2 retroviral-mediated reprogramming [26] Combinatorial Ascl1 and Dlx2 retroviral-mediated reprogramming [27] Combinatorial Nurr1 and Ngn2 AAV-mediated reprogramming [28] Combinatorial Ngn2 and Isl1 CRISPRa-mediated reprogramming [23] | |
| Other | Parkinson’s disease: mouse Alzheimer’s disease: mouse | CRISPR-CasRx [29] and Lentiviral [30] based, PTBP1 repression-mediated reprogramming MicroRNA-302/367-mediated reprograming [31] | |
| Microglia | NeuroD1 | In vitro Non-injured brain: mouse | Lentiviral-mediated reprogramming [32] Lentiviral-mediated reprogramming [32] |
| Combinatorial Approaches | Ischemic stroke: mouse Traumatic brain injury: mouse | Combinatorial Ascl1, Sox2, NeuroD1 retroviral-mediated reprogramming [25] Combinatorial OCT4OCT4, KLF4, Sox2, and c-MYC retroviral-mediated reprogramming [33] | |
| Pericyte | Combinatorial Approach | In vitro | Combinatorial Sox2 and Ascl1 retroviral-mediated reprogramming [34,35,36] |
| NG2+ Glia | NeuroD1 | Alzheimer’s disease: mouse | Retroviral-mediated reprogramming [15] |
| Other | Spinal cord injury: mouse | EGFR inhibition-mediated reprogramming [37] | |
| Fibroblast | Small Molecule and Combinatorial Approaches | In vitro | Combinatorial Oct3/4, Sox2, c-MYC, and KLF4 retroviral-mediated reprogramming [1] Combinatorial Ascl1, Brn2, Myt1l, Lmx1a, and FoxA2 lentiviral-mediated reprogramming [38] Combinatorial Ascl1, Nurr1, and Lmx1 lentiviral-mediated reprogramming [39] Small molecule-mediated reprogramming with seven-molecule cocktail [40] Combinatorial OCT4OCT4, Nanog, KLF4, c-MYC, Sox2Sox2, and hTERT mRNA plasmid transfection-mediated reprogramming [41] |
| iPSC | Sox10 | In vitro | Lentiviral-mediated Sox10-based reprogramming [42] |
| Small Molecule and Combinatorial Approaches | In vitro | Small molecule-mediated reprogramming with dual SMAD inhibitors [43] Combinatorial Ascl1, Lhx6, Dlx2, miR-9/9*-124 lentiviral-mediated reprogramming [44] Combinatorial Sox10, Olig2, and Nkx6-2 CRISPR-Cas9-mediated reprogramming [45] | |
| Other | Ascl1 | In vitro | Plasmid DNA transfection-mediated reprogramming of cochlear non-sensory epithelial cells [46] Lentiviral-mediated reprogramming of glioma cells [47] |
| Combinatorial Approaches | In vitro Glioma brain tumor: mouse | Combinatorial Ascl1, Brn2, with Ngn2 [48] or Ngn2 with Sox11 [49] lentiviral-mediated reprogramming of glioma cells Combinatorial Ngn2 and Sox11 lentiviral-mediated reprogramming of glioma cells [49] Combinatorial Ngn2 and Sox11 lentiviral-mediated reprogramming of glioma cells in mouse brain tumor [49] | |
| Other | Visual disorder: mouse | CRISPR-Cas9-mediated Nrl repression for red photoreceptor re-programming [50] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, I.H.; Roman, A.; Fellows, E.; Radha, S.; Var, S.R.; Roushdy, Z.; Borer, S.M.; Johnson, S.; Chen, O.; Borgida, J.S.; et al. Cell Reprogramming for Regeneration and Repair of the Nervous System. Biomedicines 2022, 10, 2598. https://doi.org/10.3390/biomedicines10102598
Clark IH, Roman A, Fellows E, Radha S, Var SR, Roushdy Z, Borer SM, Johnson S, Chen O, Borgida JS, et al. Cell Reprogramming for Regeneration and Repair of the Nervous System. Biomedicines. 2022; 10(10):2598. https://doi.org/10.3390/biomedicines10102598
Chicago/Turabian StyleClark, Isaac H., Alex Roman, Emily Fellows, Swathi Radha, Susanna R. Var, Zachary Roushdy, Samuel M. Borer, Samantha Johnson, Olivia Chen, Jacob S. Borgida, and et al. 2022. "Cell Reprogramming for Regeneration and Repair of the Nervous System" Biomedicines 10, no. 10: 2598. https://doi.org/10.3390/biomedicines10102598
APA StyleClark, I. H., Roman, A., Fellows, E., Radha, S., Var, S. R., Roushdy, Z., Borer, S. M., Johnson, S., Chen, O., Borgida, J. S., Steevens, A., Shetty, A., Strell, P., Low, W. C., & Grande, A. W. (2022). Cell Reprogramming for Regeneration and Repair of the Nervous System. Biomedicines, 10(10), 2598. https://doi.org/10.3390/biomedicines10102598