
Interplay between the m6A Epitranscriptome and Tumor Metabolism: Mechanisms and Therapeutic Implications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
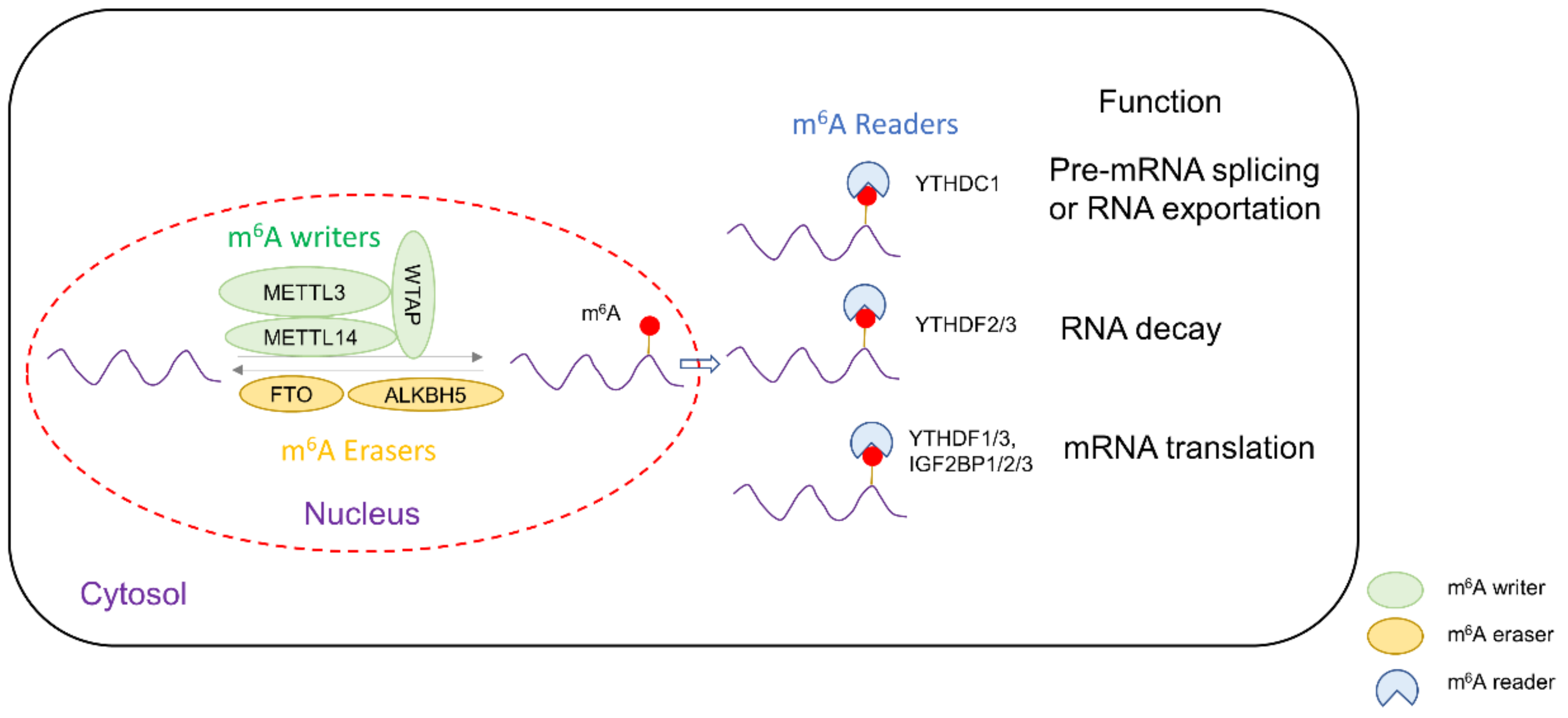
:1. Introduction
2. m6A and Glucose Metabolism in Cancer
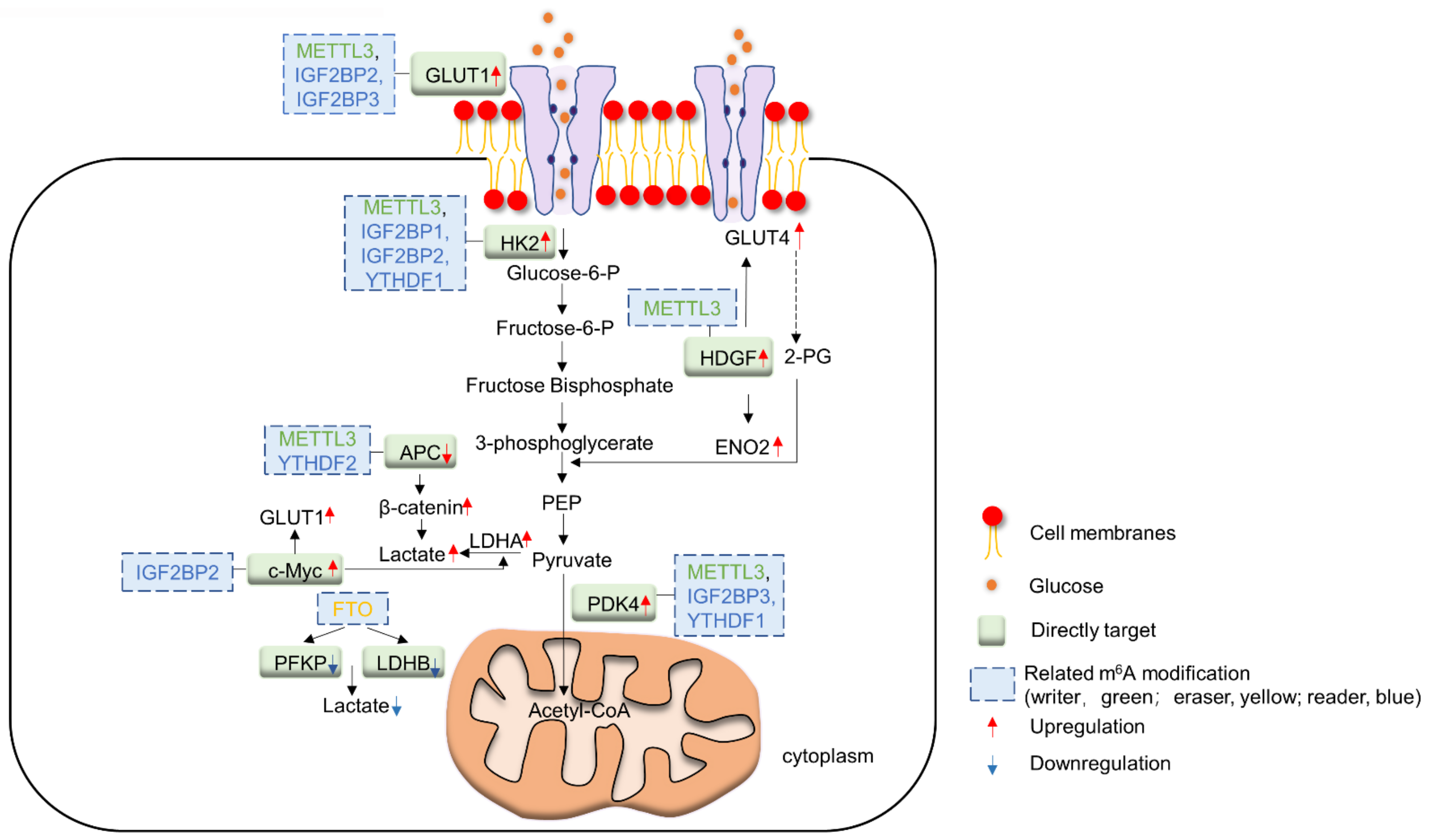
2.1. m6A Writers and Glucose Metabolism
2.2. m6A Readers and Glucose Metabolism
2.3. m6A Erasers and Glucose Metabolism
3. m6A and Lipid Metabolism in Cancer
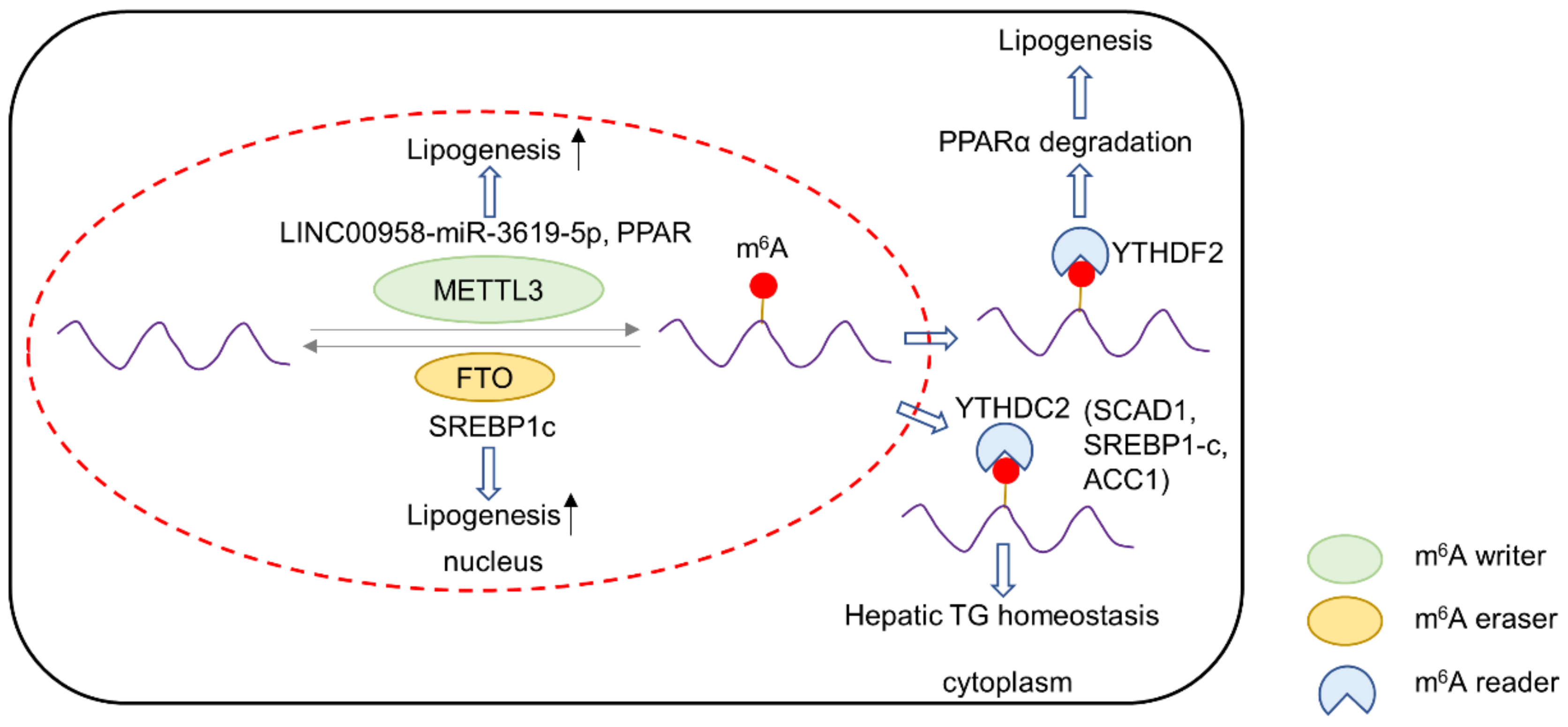
3.1. m6A Writers and Lipid Metabolism
3.2. m6A Readers and Lipid Metabolism
3.3. m6A Erasers and Lipid Metabolism
4. m6A and Amino Acid Metabolism in Cancer
4.1. m6A Writers and Methionine Metabolism
4.2. m6A Readers and Glutamine Metabolism
4.3. m6A Erasers and Glutamine Metabolism
5. Therapeutic Implications and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e14. [Google Scholar] [CrossRef] [Green Version]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N(6)-methyladenosine (m(6)A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Ramesh-Kumar, D.; Guil, S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin. Cancer Biol. 2022, in press. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [Green Version]
- Dixit, D.; Prager, B.C.; Gimple, R.C.; Poh, H.X.; Wang, Y.; Wu, Q.; Qiu, Z.; Kidwell, R.L.; Kim, L.J.Y.; Xie, Q.; et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov. 2021, 11, 480–499. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal. Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef]
- Shen, C.; Xuan, B.; Yan, T.; Ma, Y.; Xu, P.; Tian, X.; Zhang, X.; Cao, Y.; Ma, D.; Zhu, X.; et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol. Cancer 2020, 19, 72. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Yuan, L.L.; Gao, Y.; Zhou, L.M.; Yang, J.W.; Pei, Z.J. Overexpression of METTL3 associated with the metabolic status on (18)F-FDG PET/CT in patients with Esophageal Carcinoma. J. Cancer 2020, 11, 4851–4860. [Google Scholar] [CrossRef]
- Wang, W.; Shao, F.; Yang, X.; Wang, J.; Zhu, R.; Yang, Y.; Zhao, G.; Guo, D.; Sun, Y.; Wang, J.; et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N(6)-methyladenosine-dependent YTHDF binding. Nat. Commun. 2021, 12, 3803. [Google Scholar] [CrossRef]
- Li, Z.; Peng, Y.; Li, J.; Chen, Z.; Chen, F.; Tu, J.; Lin, S.; Wang, H. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat. Commun. 2020, 11, 2578. [Google Scholar] [CrossRef]
- Younes, M.; Lechago, L.V.; Somoano, J.R.; Mosharaf, M.; Lechago, J. Wide expression of the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Res. 1996, 56, 1164–1167. [Google Scholar]
- Chen, H.; Gao, S.; Liu, W.; Wong, C.C.; Wu, J.; Wu, J.; Liu, D.; Gou, H.; Kang, W.; Zhai, J.; et al. RNA N(6)-Methyladenosine Methyltransferase METTL3 Facilitates Colorectal Cancer by Activating the m(6)A-GLUT1-mTORC1 Axis and Is a Therapeutic Target. Gastroenterology 2021, 160, 1284–1300.e16. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, X.; Li, L.; Gao, Z.; Su, X.; Ji, M.; Liu, J. N(6)-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell. Death Dis. 2020, 11, 911. [Google Scholar] [CrossRef]
- Fry, N.J.; Law, B.A.; Ilkayeva, O.R.; Holley, C.L.; Mansfield, K.D. N(6)-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA 2017, 23, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Li, K.J.; Feng, J.X.; Liu, G.J.; Feng, Y.L. Blocking the IGF2BP1-promoted glucose metabolism of colon cancer cells via direct de-stabilizing mRNA of the LDHA enhances anticancer effects. Mol. Ther. Nucleic Acids 2021, 23, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, J.H.; Wu, Q.N.; Jin, Y.; Wang, D.S.; Chen, Y.X.; Liu, J.; Luo, X.J.; Meng, Q.; Pu, H.Y.; et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol. Cancer 2019, 18, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Liu, X.; Zhou, S.; Li, W.; Liu, C.; Chadwick, M.; Qian, C. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. 2019, 450, 63–75. [Google Scholar] [CrossRef]
- Zhai, S.; Xu, Z.; Xie, J.; Zhang, J.; Wang, X.; Peng, C.; Li, H.; Chen, H.; Shen, B.; Deng, X. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene 2021, 40, 277–291. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, Q.; Pang, W.; Hou, L.; Liang, Y.; Han, X.; Luo, X.; Wang, P.; Zhang, X.; Li, L.; et al. YTHDC1-mediated augmentation of miR-30d in repressing pancreatic tumorigenesis via attenuation of RUNX1-induced transcriptional activation of Warburg effect. Cell Death Differ. 2021, 28, 3105–3124. [Google Scholar] [CrossRef]
- Niu, Y.; Lin, Z.; Wan, A.; Sun, L.; Yan, S.; Liang, H.; Zhan, S.; Chen, D.; Bu, X.; Liu, P.; et al. Loss-of-Function Genetic Screening Identifies Aldolase A as an Essential Driver for Liver Cancer Cell Growth Under Hypoxia. Hepatology 2021, 74, 1461–1479. [Google Scholar] [CrossRef]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939.e9. [Google Scholar] [CrossRef]
- Yang, X.; Shao, F.; Guo, D.; Wang, W.; Wang, J.; Zhu, R.; Gao, Y.; He, J.; Lu, Z. WNT/beta-catenin-suppressed FTO expression increases m(6)A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 2021, 12, 462. [Google Scholar] [CrossRef]
- Yu, H.; Yang, X.; Tang, J.; Si, S.; Zhou, Z.; Lu, J.; Han, J.; Yuan, B.; Wu, Q.; Lu, Q.; et al. ALKBH5 Inhibited Cell Proliferation and Sensitized Bladder Cancer Cells to Cisplatin by m6A-CK2alpha-Mediated Glycolysis. Mol. Ther. Nucleic Acids 2021, 23, 27–41. [Google Scholar] [CrossRef]
- Li, N.; Kang, Y.; Wang, L.; Huff, S.; Tang, R.; Hui, H.; Agrawal, K.; Gonzalez, G.M.; Wang, Y.; Patel, S.P.; et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 20159–20170. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ohsugi, M.; Sasako, T.; Awazawa, M.; Umehara, T.; Iwane, A.; Kobayashi, N.; Okazaki, Y.; Kubota, N.; Suzuki, R.; et al. The RNA Methyltransferase Complex of WTAP, METTL3, and METTL14 Regulates Mitotic Clonal Expansion in Adipogenesis. Mol. Cell. Biol. 2018, 38, e00116–e00118. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Chen, Z.; Gao, W.; Zhang, Y.; Wang, J.; Wang, J.; Cao, M.; Cai, J.; Wu, J.; Wang, X. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J. Hematol. Oncol. 2020, 13, 5. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Ma, L.L.; Xu, Y.Q.; Wang, B.H.; Li, S.M. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of Fasn mRNA and promoting fatty acid metabolism. Biochem. Biophys. Res. Commun. 2019, 518, 120–126. [Google Scholar] [CrossRef]
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m(6)A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e4. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, S.; Nakano, M.; Fukami, T.; Nakajima, M. m(6)A modification impacts hepatic drug and lipid metabolism properties by regulating carboxylesterase 2. Biochem. Pharmacol. 2021, 193, 114766. [Google Scholar] [CrossRef]
- Sun, D.; Zhao, T.; Zhang, Q.; Wu, M.; Zhang, Z. Fat mass and obesity-associated protein regulates lipogenesis via m(6) A modification in fatty acid synthase mRNA. Cell. Biol. Int. 2021, 45, 334–344. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, C.; Xu, L.; Yuan, Y.; Zhao, J.; Zhao, W.; Chen, Y.; Qiu, J.; Meng, M.; Zheng, Y.; et al. N(6)-Methyladenosine Reader Protein YT521-B Homology Domain-Containing 2 Suppresses Liver Steatosis by Regulation of mRNA Stability of Lipogenic Genes. Hepatology 2021, 73, 91–103. [Google Scholar] [CrossRef]
- Song, T.; Yang, Y.; Wei, H.; Xie, X.; Lu, J.; Zeng, Q.; Peng, J.; Zhou, Y.; Jiang, S.; Peng, J. Zfp217 mediates m6A mRNA methylation to orchestrate transcriptional and post-transcriptional regulation to promote adipogenic differentiation. Nucleic Acids Res. 2019, 47, 6130–6144. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Wu, R.; Liu, Y.; Yao, Y.; Zhao, Y.; Bi, Z.; Jiang, Q.; Liu, Q.; Cai, M.; Wang, F.; Wang, Y.; et al. FTO regulates adipogenesis by controlling cell cycle progression via m(6)A-YTHDF2 dependent mechanism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Merkestein, M.; Laber, S.; McMurray, F.; Andrew, D.; Sachse, G.; Sanderson, J.; Li, M.; Usher, S.; Sellayah, D.; Ashcroft, F.M.; et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat. Commun. 2015, 6, 6792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Chen, X.; Cheng, S.; Shu, L.; Yan, M.; Yao, L.; Wang, B.; Huang, S.; Zhou, L.; Yang, Z.; et al. FTO promotes SREBP1c maturation and enhances CIDEC transcription during lipid accumulation in HepG2 cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Chiang, P.K.; Gordon, R.K.; Tal, J.; Zeng, G.C.; Doctor, B.P.; Pardhasaradhi, K.; McCann, P.P. S-Adenosylmethionine and methylation. FASEB J. 1996, 10, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol 2017, 45, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, H.; Kashyap, S.; Cobo-Stark, P.; Flaten, A.; Chang, C.M.; Hajarnis, S.; Hein, K.Z.; Lika, J.; Warner, G.M.; Espindola-Netto, J.M.; et al. A methionine-Mettl3-N(6)-methyladenosine axis promotes polycystic kidney disease. Cell Metab. 2021, 33, 1234–1247.e7. [Google Scholar] [CrossRef]
- Shima, H.; Matsumoto, M.; Ishigami, Y.; Ebina, M.; Muto, A.; Sato, Y.; Kumagai, S.; Ochiai, K.; Suzuki, T.; Igarashi, K. S-Adenosylmethionine Synthesis Is Regulated by Selective N(6)-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep. 2017, 21, 3354–3363. [Google Scholar] [CrossRef] [Green Version]
- Watabe, E.; Togo-Ohno, M.; Ishigami, Y.; Wani, S.; Hirota, K.; Kimura-Asami, M.; Hasan, S.; Takei, S.; Fukamizu, A.; Suzuki, Y.; et al. m(6)A-mediated alternative splicing coupled with nonsense-mediated mRNA decay regulates SAM synthetase homeostasis. EMBO J. 2021, 40, e106434. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Wong, C.C.; Xu, J.; Bian, X.; Wu, J.L.; Kang, W.; Qian, Y.; Li, W.; Chen, H.; Gou, H.; Liu, D.; et al. In Colorectal Cancer Cells with Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Increase WNT Signaling, Stemness, and Drug Resistance. Gastroenterology 2020, 159, 2163–2180.e6. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Li, X.; Xu, J.; Kang, W.; Tong, J.H.; To, K.F.; Jin, Y.; Li, W.; Chen, H.; et al. SLC25A22 Promotes Proliferation and Survival of Colorectal Cancer Cells with KRAS Mutations and Xenograft Tumor Progression in Mice via Intracellular Synthesis of Aspartate. Gastroenterology 2016, 151, 945–960.e6. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Gao, S.; Zeng, Y.; Zhu, L.; Mo, Y.; Wong, C.C.; Bao, Y.; Su, P.; Zhai, J.; Wang, L.; et al. N6-Methyladenosine Reader YTHDF1 Promotes ARHGEF2 Translation and RhoA Signaling in Colorectal Cancer. Gastroenterology 2022, 162, 1183–1196. [Google Scholar] [CrossRef]
- Chen, P.; Liu, X.Q.; Lin, X.; Gao, L.Y.; Zhang, S.; Huang, X. Targeting YTHDF1 effectively re-sensitizes cisplatin-resistant colon cancer cells by modulating GLS-mediated glutamine metabolism. Mol. Ther. Oncolytics 2021, 20, 228–239. [Google Scholar] [CrossRef]
- Han, S.; Zhu, L.; Zhu, Y.; Meng, Y.; Li, J.; Song, P.; Yousafzai, N.A.; Feng, L.; Chen, M.; Wang, Y.; et al. Targeting ATF4-dependent pro-survival autophagy to synergize glutaminolysis inhibition. Theranostics 2021, 11, 8464–8479. [Google Scholar] [CrossRef]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Thakkar, K.N.; Zhao, H.; Broughton, J.; Li, Y.; Seoane, J.A.; Diep, A.N.; Metzner, T.J.; von Eyben, R.; Dill, D.L.; et al. The m(6)A RNA demethylase FTO is a HIF-independent synthetic lethal partner with the VHL tumor suppressor. Proc. Natl. Acad. Sci. USA 2020, 117, 21441–21449. [Google Scholar] [CrossRef]
- Yankova, E.; Blackaby, W.; Albertella, M.; Rak, J.; De Braekeleer, E.; Tsagkogeorga, G.; Pilka, E.S.; Aspris, D.; Leggate, D.; Hendrick, A.G.; et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 2021, 593, 597–601. [Google Scholar] [CrossRef]
- Li, Q.; Huang, Y.; Liu, X.; Gan, J.; Chen, H.; Yang, C.G. Rhein Inhibits AlkB Repair Enzymes and Sensitizes Cells to Methylated DNA Damage. J. Biol. Chem. 2016, 291, 11083–11093. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384. [Google Scholar] [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.e10. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fong, A.S.; Pan, Y.; Yu, J.; Wong, C.C. Interplay between the m6A Epitranscriptome and Tumor Metabolism: Mechanisms and Therapeutic Implications. Biomedicines 2022, 10, 2589. https://doi.org/10.3390/biomedicines10102589
Fong AS, Pan Y, Yu J, Wong CC. Interplay between the m6A Epitranscriptome and Tumor Metabolism: Mechanisms and Therapeutic Implications. Biomedicines. 2022; 10(10):2589. https://doi.org/10.3390/biomedicines10102589
Chicago/Turabian StyleFong, Asa Sergei, Yasi Pan, Jun Yu, and Chi Chun Wong. 2022. "Interplay between the m6A Epitranscriptome and Tumor Metabolism: Mechanisms and Therapeutic Implications" Biomedicines 10, no. 10: 2589. https://doi.org/10.3390/biomedicines10102589
APA StyleFong, A. S., Pan, Y., Yu, J., & Wong, C. C. (2022). Interplay between the m6A Epitranscriptome and Tumor Metabolism: Mechanisms and Therapeutic Implications. Biomedicines, 10(10), 2589. https://doi.org/10.3390/biomedicines10102589