
Exogenous Antioxidants in Remyelination and Skeletal Muscle Recovery

 and
and
Abstract
1. Introduction
2. Autoimmune and Inflammatory Demyelination
2.1. Role of Sphingolipids and ROS
2.2. Inflammation in Demyelinating Diseases
3. Skeletal Muscle Damage and Oxidative Stress Demyelination Related
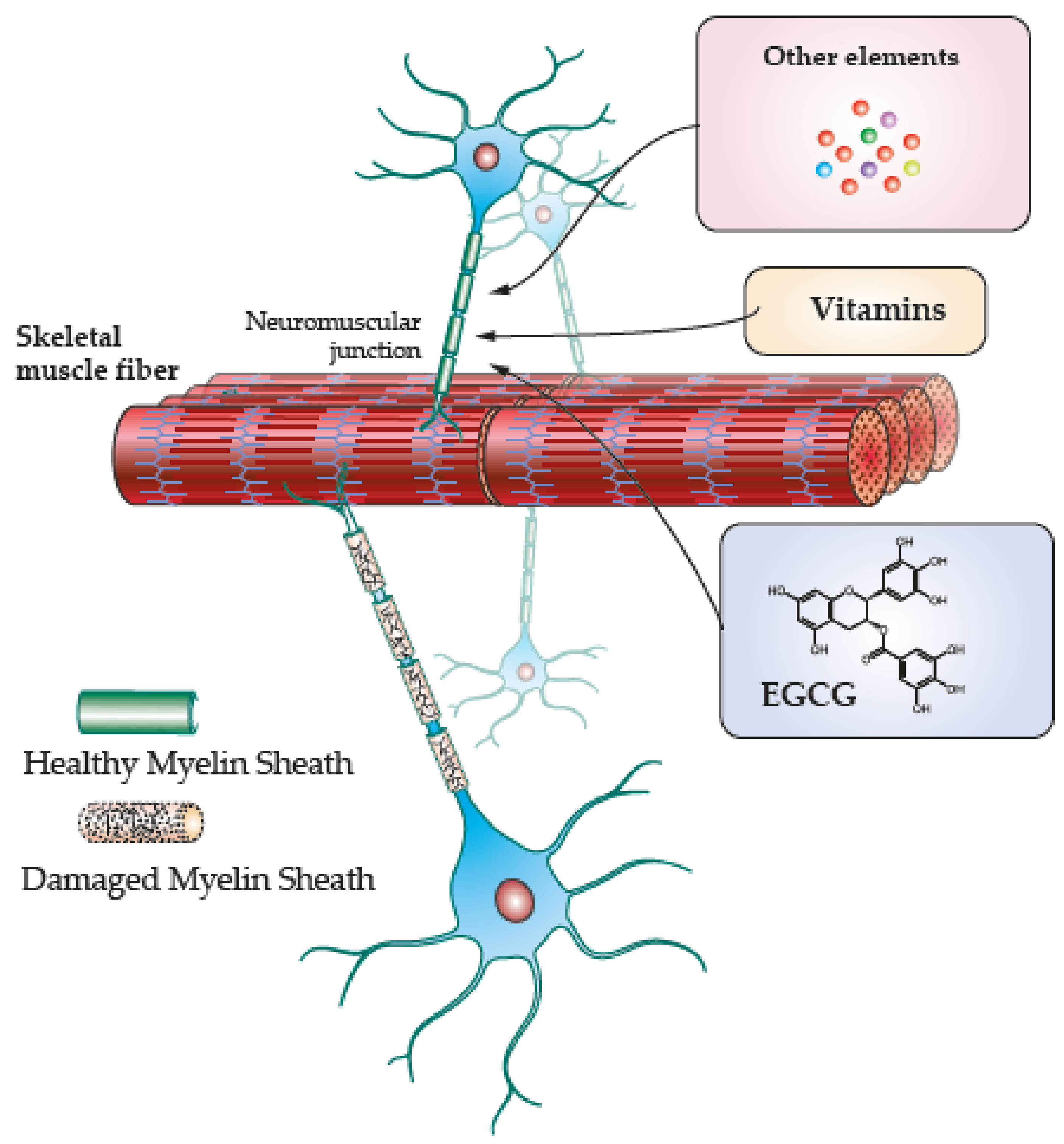
4. Skeletal Muscle Regeneration along Antioxidant Induced Remyelination
4.1. Curcumin
4.2. Flavonoids
4.3. EGCG
4.4. Vitamins and Other Elements

{kind=link}
{kind=link}
| Compound | Type | Effect on Muscle | Effect on Brain/Neurons | References |
|---|---|---|---|---|
| Vitamin C | Ascorbic acid | Antioxidant | Peripheral nerve development Antioxidant | [17] |
| Quercetin | Flavonoid | Recovery of neuromuscular function. Reduction in skeletal muscle atrophy. | Antiinflammatory neuroprotection | [19] |
| Baicalin | Flavone glicoside | Reduce skeletal muscle damage | Oligodendrocyte proliferation, differentiation. Improve remyelination, activation of PGC1a | [82,83] |
| Icariin | Flavonoid | Angiogenesis, tendon bone healing. | Myelin restoration, oligodendrocyte maturation, BDNF increase. | [84] |
| EGCG | Polyphenol | Increase myotubes number and length Myogenic differentiation Increase muscle area Increase antioxidative production. Reduction lactate and stable carbohydrate oxidation | Increase myelin sheath thickness Nociceptive recovery Improvements in posture | [85,86,87,88,89,90,91,92,93,94,95,96,97,98] |
| Vitamin E/ | Tocopherol/ | Antioxidant Muscle recovery | Antioxidant effects reduced apoptosis, increase remyelination. Improvements in nerve and motor function. | [99,100,101] |
| Vitamin D3 | Cholecalciferol | Antioxidant effects Reduced apoptosis, increase remyelination | ||
| Selenium | Antioxidant, microglial inhibition, increased remyelination | [102,103,104,105,106] |
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADEM | Acute disseminated encephalomyelitis |
| AQP4 | Aquaporin 4 |
| BDNF | Brain-derived 590 neurotrophic factor |
| CCL | Chemokine |
| CD3 | Cluster of differentiation 3 |
| C1P | Ceramide1-phosphate |
| COX-2 | Cyclooxygenase 2 |
| CSF-1 | Colony stimulating factor |
| EGCG | Epigallocatechin Gallate |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| GFAP | Glial acidic fibrillary protein |
| HIF-1 | Hypoxia inducible factor-1 |
| IGF-1 | Insulin-like growth factor 1 |
| IL | Interleukin |
| JAK-STAT | JAK (Janus Kinase)-STAT signaling pathway |
| MAG | Myelin-associated glycoproteins |
| MDEM | Multiphasic disseminated encephalomyelitis |
| MHC-II | Major 301 histocompatibility-II Complex |
| Myrf | Myelin regulatory factor |
| MOG | Myelin oligodendrocyte glycoprotein |
| NF-kB | Nuclear Factor type KB |
| Nrf2 | Nuclear factor E2-related factor |
| ORMDL3 | ORMDL (Orm1-like3) sphingolipid biosynthesis Regulator 3 |
| PLC | Phospholipase C |
| PGE2 | Prostaglandin E2 |
| PGC1a | Peroxisome proliferator-activated receptor gamma co-activator 1-alpha |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| ROS | Reactive Oxygen Species |
| SC | Schwann cells |
| SMase | Sphingomyelinase |
| S1P | Sphingosine1-phosphate |
| SOD | Superoxide dismutase |
| TNF | Tumoral necrosis factor |
| TGF-β | Transforming growth factor beta |
References
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Mahad, D.J. Mitochondrial changes associated with demyelination: Consequences for axonal integrity. Mitochondrion 2012, 12, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxid. Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Gui, L.N.; Liu, Y.Y.; Shi, S.; Cheng, Y. Oxidative Stress Marker Aberrations in Multiple Sclerosis: A Meta-Analysis Study. Front. Neurosci. 2020, 14, 823. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F. Sphingolipids in inflammation: Pathological implications and potential therapeutic targets. Br. J. Pharmacol. 2009, 158, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Myelin damage and repair in pathologic CNS: Challenges and prospects. Front. Mol. Neurosci. 2015, 8, 35. [Google Scholar] [CrossRef]
- Yin, L.; Li, N.; Jia, W.; Wang, N.; Liang, M.; Yang, X.; Du, G. Skeletal muscle atrophy: From mechanisms to treatments. Pharmacol. Res. 2021, 172, 105807. [Google Scholar] [CrossRef] [PubMed]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, R.; Xu, L.; Wan, Q.; Zhu, J.; Gu, J.; Huang, Z.; Ma, W.; Shen, M.; Ding, F.; et al. Microarray Analysis of Gene Expression Provides New Insights Into Denervation-Induced Skeletal Muscle Atrophy. Front. Physiol. 2019, 10, 1298. [Google Scholar] [CrossRef]
- Ahmad, K.; Shaikh, S.; Ahmad, S.S.; Lee, E.J.; Choi, I. Cross-Talk Between Extracellular Matrix and Skeletal Muscle: Implications for Myopathies. Front. Pharmacol. 2020, 11, 142. [Google Scholar] [CrossRef]
- Bhagavati, S. Autoimmune Disorders of the Nervous System: Pathophysiology, Clinical Features, and Therapy. Front. Neurol. 2021, 12, 664664. [Google Scholar] [CrossRef]
- Chu, X.L.; Song, X.Z.; Li, Q.; Li, Y.R.; He, F.; Gu, X.S.; Ming, D. Basic mechanisms of peripheral nerve injury and treatment via electrical stimulation. Neural. Regen. Res. 2022, 17, 2185–2193. [Google Scholar] [CrossRef]
- Simon, J.P.; Marie, I.; Jouen, F.; Boyer, O.; Martinet, J. Autoimmune Myopathies: Where Do We Stand? Front. Immunol. 2016, 7, 234. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Podratz, J.L.; Rodriguez, E.H.; Windebank, A.J. Antioxidants are necessary for myelination of dorsal root ganglion neurons, in vitro. Glia 2004, 45, 54–58. [Google Scholar] [CrossRef]
- Dong, L.; Li, R.; Li, D.; Wang, B.; Lu, Y.; Li, P.; Yu, F.; Jin, Y.; Ni, X.; Wu, Y.; et al. FGF10 Enhances Peripheral Nerve Regeneration via the Preactivation of the PI3K/Akt Signaling-Mediated Antioxidant Response. Front. Pharm. 2019, 10, 1224. [Google Scholar] [CrossRef] [PubMed]
- Simioni, C.; Zauli, G.; Martelli, A.M.; Vitale, M.; Sacchetti, G.; Gonelli, A.; Neri, L.M. Oxidative stress: Role of physical exercise and antioxidant nutraceuticals in adulthood and aging. Oncotarget 2018, 9, 17181–17198. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lin, Q.; Liang, Y. Plant-Derived Antioxidants Protect the Nervous System From Aging by Inhibiting Oxidative Stress. Front. Aging Neurosci. 2020, 12, 209. [Google Scholar] [CrossRef] [PubMed]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 3887. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jin, H.K.; Bae, J.S. Sphingolipids in neuroinflammation: A potential target for diagnosis and therapy. BMB Rep. 2020, 53, 28–34. [Google Scholar] [CrossRef]
- Latov, C.B.T.H.B.W.T.N. Chronic Inflammatory Demyelinating Polineuropathy. Neuromusc. Disord. 1996, 6, 311–325. [Google Scholar]
- Tidball, J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011, 1, 2029–2062. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Fischer, M.T.; Frischer, J.M.; Bauer, J.; Hoftberger, R.; Botond, G.; Esterbauer, H.; Binder, C.J.; Witztum, J.L.; Lassmann, H. Oxidative damage in multiple sclerosis lesions. Brain 2011, 134, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Sinyuk, M.; Jenkins, J.E., 3rd; Psenicka, M.W.; Williams, J.L. The impact of regional astrocyte interferon-gamma signaling during chronic autoimmunity: A novel role for the immunoproteasome. J. Neuroinflammation 2020, 17, 184. [Google Scholar] [CrossRef] [PubMed]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Sphingolipids in inflammation: Roles and implications. Curr. Mol. Med. 2004, 4, 405–418. [Google Scholar] [CrossRef]
- Bugajev, V.; Paulenda, T.; Utekal, P.; Mrkacek, M.; Halova, I.; Kuchar, L.; Kuda, O.; Vavrova, P.; Schuster, B.; Fuentes-Liso, S.; et al. Crosstalk between ORMDL3, serine palmitoyltransferase, and 5-lipoxygenase in the sphingolipid and eicosanoid metabolic pathways. J. Lipid Res. 2021, 62, 100121. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.A.; Majumder, S.; Zhu, H.; Lee, Y.T.; Kono, M.; Li, C.; Khanna, C.; Blain, H.; Schwartz, R.; Huso, V.L.; et al. The Ormdl genes regulate the sphingolipid synthesis pathway to ensure proper myelination and neurologic function in mice. eLife 2019, 8, e51067. [Google Scholar] [CrossRef]
- Kim, S.; Steelman, A.J.; Zhang, Y.; Kinney, H.C.; Li, J. Aberrant upregulation of astroglial ceramide potentiates oligodendrocyte injury. Brain Pathol. 2012, 22, 41–57. [Google Scholar] [CrossRef]
- Rodriguez, Y.; Vatti, N.; Ramirez-Santana, C.; Chang, C.; Mancera-Paez, O.; Gershwin, M.E.; Anaya, J.M. Chronic inflammatory demyelinating polyneuropathy as an autoimmune disease. J. Autoimmun. 2019, 102, 8–37. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Park, H.T.; Kim, Y.H.; Lee, K.E.; Kim, J.K. Behind the pathology of macrophage-associated demyelination in inflammatory neuropathies: Demyelinating Schwann cells. Cell. Mol. Life Sci. CMLS 2020, 77, 2497–2506. [Google Scholar] [CrossRef]
- Yadav, A.; Huang, T.C.; Chen, S.H.; Ramasamy, T.S.; Hsueh, Y.Y.; Lin, S.P.; Lu, F.I.; Liu, Y.H.; Wu, C.C. Sodium phenylbutyrate inhibits Schwann cell inflammation via HDAC and NFkappaB to promote axonal regeneration and remyelination. J. Neuroinflammation 2021, 18, 238. [Google Scholar] [CrossRef]
- Kazamel, M.; Stino, A.M.; Smith, A.G. Metabolic syndrome and peripheral neuropathy. Muscle Nerve 2021, 63, 285–293. [Google Scholar] [CrossRef]
- Zilliox, L.A. Diabetes and Peripheral Nerve Disease. Clin. Geriatr. Med. 2021, 37, 253–267. [Google Scholar] [CrossRef]
- Perandini, L.A.; Chimin, P.; Lutkemeyer, D.D.S.; Camara, N.O.S. Chronic inflammation in skeletal muscle impairs satellite cells function during regeneration: Can physical exercise restore the satellite cell niche? FEBS J. 2018, 285, 1973–1984. [Google Scholar] [CrossRef]
- Hoftberger, R.; Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. Handb. Clin. Neurol. 2017, 145, 263–283. [Google Scholar] [CrossRef]
- Dufresne, S.S.; Frenette, J.; Dumont, N.A. Inflammation and muscle regeneration, a double-edged sword. Med. Sci. M/S 2016, 32, 591–597. [Google Scholar] [CrossRef]
- Superstein, R.J.B.D.S. Guillain-BarrC Syndrome and Chronic Inflammatory Demyelinating Polyneuropathy. Semin. Neurol. 1998, 18, 1–13. [Google Scholar]
- Mayo, L.; Quintana, F.J.; Weiner, H.L. The innate immune system in demyelinating disease. Immunol. Rev. 2012, 248, 170–187. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Mirshafiey, A.; Mohsenzadegan, M. Antioxidant therapy in multiple sclerosis. Immunopharmacol. Immunotoxicol. 2009, 31, 13–29. [Google Scholar] [CrossRef]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weissflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef]
- Nellessen, A.; Nyamoya, S.; Zendedel, A.; Slowik, A.; Wruck, C.; Beyer, C.; Fragoulis, A.; Clarner, T. Nrf2 deficiency increases oligodendrocyte loss, demyelination, neuroinflammation and axonal damage in an MS animal model. Metab. Brain Dis. 2020, 35, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Johnson, D.; Johnson, J.A. Deletion of Nrf2 impairs functional recovery, reduces clearance of myelin debris and decreases axonal remyelination after peripheral nerve injury. Neurobiol. Dis. 2013, 54, 329–338. [Google Scholar] [CrossRef]
- Hu, W.; Lucchinetti, C.F. The pathological spectrum of CNS inflammatory demyelinating diseases. Semin. Immunopathol. 2009, 31, 439–453. [Google Scholar] [CrossRef]
- Hardy, T.A.; Reddel, S.W.; Barnett, M.H.; Palace, J.; Lucchinetti, C.F.; Weinshenker, B.G. Atypical inflammatory demyelinating syndromes of the CNS. Lancet Neurol. 2016, 15, 967–981. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.; Duan, Z.; Chen, K.; Liu, Z.; Zhang, L.; Yao, D.; Li, B. The Effects of Co-transplantation of Olfactory Ensheathing Cells and Schwann Cells on Local Inflammation Environment in the Contused Spinal Cord of Rats. Mol. Neurobiol. 2017, 54, 943–953. [Google Scholar] [CrossRef]
- Trias, E.; Kovacs, M.; King, P.H.; Si, Y.; Kwon, Y.; Varela, V.; Ibarburu, S.; Moura, I.C.; Hermine, O.; Beckman, J.S.; et al. Schwann cells orchestrate peripheral nerve inflammation through the expression of CSF1, IL-34, and SCF in amyotrophic lateral sclerosis. Glia 2020, 68, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.J.; Hammock, B.; Cortopassi, G. Frataxin deficiency induces Schwann cell inflammation and death. Biochim. Et Biophys. Acta 2009, 1792, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Liu, H.; Kikuchi, S.; Myers, R.R.; Shubayev, V.I. Immediate anti-tumor necrosis factor-alpha (etanercept) therapy enhances axonal regeneration after sciatic nerve crush. J. Neurosci. Res. 2010, 88, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Hartlehnert, M.; Derksen, A.; Hagenacker, T.; Kindermann, D.; Schafers, M.; Pawlak, M.; Kieseier, B.C.; Meyer Zu Horste, G. Schwann cells promote post-traumatic nerve inflammation and neuropathic pain through MHC class II. Sci. Rep. 2017, 7, 12518. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Zhang, H. Extracellular matrix: An important regulator of cell functions and skeletal muscle development. Cell Biosci. 2021, 11, 65. [Google Scholar] [CrossRef]
- Rosero Salazar, D.H.; Elvira Flórez, L.J. Image analysis in Gomori’s trichrome stain of skeletal muscles subjected to ischemia and reperfusion injury. Explor. Anim. Med. Res. 2016, 6, 15–25. [Google Scholar]
- Salazar, D.H.R.; Monsalve, L.S. Digital image analysis of striated skeletal muscle tissue injury during reperfusion after induced ischemia. Proc. SPIE 2015, 9287, 92870I. [Google Scholar] [CrossRef]
- Purslow, P.P. Muscle fascia and force transmission. J. Bodyw. Mov. 2010, 14, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Rosero Salazar, D.H.; Carvajal Monroy, P.L.; Wagener, F.; Von den Hoff, J.W. Orofacial Muscles: Embryonic Development and Regeneration after Injury. J. Dent. Res. 2020, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef]
- Pruller, J.; Mannhardt, I.; Eschenhagen, T.; Zammit, P.S.; Figeac, N. Satellite cells delivered in their niche efficiently generate functional myotubes in three-dimensional cell culture. PLoS ONE 2018, 13, e0202574. [Google Scholar] [CrossRef]
- Chawla, J. Stepwise approach to myopathy in systemic disease. Front. Neurol. 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Nagy, H.; Veerapaneni, K.D. Myopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar] [PubMed]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.A.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.; S, A.S.; Xiao, L.; Teuscher, C.; et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 2016, 5, e18246. [Google Scholar] [CrossRef] [PubMed]
- Abu, R.; Yu, L.; Kumar, A.; Gao, L.; Kumar, V. A Quantitative Proteomics Approach to Gain Insight into NRF2-KEAP1 Skeletal Muscle System and Its Cysteine Redox Regulation. Genes 2021, 12, 1655. [Google Scholar] [CrossRef]
- Vallee, A.; Lecarpentier, Y. TGF-beta in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, J.R.; Hoffman, D.B.; Corona, B.T.; Greising, S.M. Secondary denervation is a chronic pathophysiologic sequela of volumetric muscle loss. J. Appl. Physiol. (1985) 2021, 130, 1614–1625. [Google Scholar] [CrossRef]
- Greising, S.M.; Warren, G.L.; Southern, W.M.; Nichenko, A.S.; Qualls, A.E.; Corona, B.T.; Call, J.A. Early rehabilitation for volumetric muscle loss injury augments endogenous regenerative aspects of muscle strength and oxidative capacity. BMC Musculoskelet. Disord. 2018, 19, 173. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Hwang, S.M.; Gomes, A.V. Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol. Cell. Biol. 2014, 34, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef]
- Ribot, C.; Soler, C.; Chartier, A.; Al Hayek, S.; Nait-Saidi, R.; Barbezier, N.; Coux, O.; Simonelig, M. Activation of the ubiquitin-proteasome system contributes to oculopharyngeal muscular dystrophy through muscle atrophy. PLoS Genet. 2022, 18, e1010015. [Google Scholar] [CrossRef]
- Haberecht-Muller, S.; Kruger, E.; Fielitz, J. Out of Control: The Role of the Ubiquitin Proteasome System in Skeletal Muscle during Inflammation. Biomolecules 2021, 11, 1327. [Google Scholar] [CrossRef] [PubMed]
- Pierucci, F.; Frati, A.; Battistini, C.; Penna, F.; Costelli, P.; Meacci, E. Control of Skeletal Muscle Atrophy Associated to Cancer or Corticosteroids by Ceramide Kinase. Cancers 2021, 13, 3285. [Google Scholar] [CrossRef] [PubMed]
- De Larichaudy, J.; Zufferli, A.; Serra, F.; Isidori, A.M.; Naro, F.; Dessalle, K.; Desgeorges, M.; Piraud, M.; Cheillan, D.; Vidal, H.; et al. TNF-alpha- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet. Muscle 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- McNally, B.D.; Ashley, D.F.; Hanschke, L.; Daou, H.N.; Watt, N.T.; Murfitt, S.A.; MacCannell, A.D.V.; Whitehead, A.; Bowen, T.S.; Sanders, F.W.B.; et al. Long-chain ceramides are cell non-autonomous signals linking lipotoxicity to endoplasmic reticulum stress in skeletal muscle. Nat. Commun. 2022, 13, 1748. [Google Scholar] [CrossRef]
- Chen, C.Z.; Neumann, B.; Forster, S.; Franklin, R.J.M. Schwann cell remyelination of the central nervous system: Why does it happen and what are the benefits? Open Biol. 2021, 11, 200352. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.W.; Agarwal, A.; Smith, M.D.; Khuder, S.S.; Baxi, E.G.; Thomas, A.G.; Rojas, C.; Moniruzzaman, M.; Slusher, B.S.; Bergles, D.E.; et al. Inhibition of neutral sphingomyelinase 2 promotes remyelination. Sci. Adv. 2020, 6, eaba5210. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.J.; Manesh, S.B.; Hilton, B.J.; Assinck, P.; Liu, J.; Moulson, A.; Plemel, J.R.; Tetzlaff, W. Locomotor recovery following contusive spinal cord injury does not require oligodendrocyte remyelination. Nat. Commun. 2018, 9, 3066. [Google Scholar] [CrossRef] [PubMed]
- Luk, H.Y.; Appell, C.; Chyu, M.C.; Chen, C.H.; Wang, C.Y.; Yang, R.S.; Shen, C.L. Impacts of Green Tea on Joint and Skeletal Muscle Health: Prospects of Translational Nutrition. Antioxidants 2020, 9, 1050. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Plumitallo, C.; De Nuccio, C.; Visentin, S.; Minghetti, L. Curcumin promotes oligodendrocyte differentiation and their protection against TNF-alpha through the activation of the nuclear receptor PPAR-gamma. Sci. Rep. 2021, 11, 4952. [Google Scholar] [CrossRef]
- Caillaud, M.; Chantemargue, B.; Richard, L.; Vignaud, L.; Favreau, F.; Faye, P.A.; Vignoles, P.; Sturtz, F.; Trouillas, P.; Vallat, J.M.; et al. Local low dose curcumin treatment improves functional recovery and remyelination in a rat model of sciatic nerve crush through inhibition of oxidative stress. Neuropharmacology 2018, 139, 98–116. [Google Scholar] [CrossRef]
- Ai, R.S.; Xing, K.; Deng, X.; Han, J.J.; Hao, D.X.; Qi, W.H.; Han, B.; Yang, Y.N.; Li, X.; Zhang, Y. Baicalin Promotes CNS Remyelination via PPARgamma Signal Pathway. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1142. [Google Scholar] [CrossRef] [PubMed]
- Silva, V.; Segura-Aguilar, J. State and perspectives on flavonoid neuroprotection against aminochrome-induced neurotoxicity. Neural. Regen. Res. 2021, 16, 1797–1798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, L.; Zheng, N.; Zhang, L.; Liu, J.; Liang, W.; Wang, Q. Icariin enhances remyelination process after acute demyelination induced by cuprizone exposure. Brain Res. Bull. 2017, 130, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.R.; Kim, K.M.; Byun, M.R.; Hwang, J.H.; Park, J.I.; Oh, H.T.; Jeong, M.G.; Hwang, E.S.; Hong, J.H. (-)-Epigallocatechin-3-gallate stimulates myogenic differentiation through TAZ activation. Biochem. Biophys. Res. Commun. 2017, 486, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Liu, H.W.; Chan, Y.C.; Hu, S.H.; Liu, M.Y.; Chang, S.J. The green tea polyphenol epigallocatechin-3-gallate attenuates age-associated muscle loss via regulation of miR-486-5p and myostatin. Arch. Biochem. Biophys. 2020, 692, 108511. [Google Scholar] [CrossRef]
- Jeong, M.G.; Kim, H.K.; Hwang, E.S. The essential role of TAZ in normal tissue homeostasis. Arch. Pharm. Res. 2021, 44, 253–262. [Google Scholar] [CrossRef]
- Wang, Q.; McPherron, A.C. Myostatin inhibition induces muscle fibre hypertrophy prior to satellite cell activation. J. Physiol. 2012, 590, 2151–2165. [Google Scholar] [CrossRef]
- Li, Z.B.; Kollias, H.D.; Wagner, K.R. Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem. 2008, 283, 19371–19378. [Google Scholar] [CrossRef]
- Nakae, Y.; Hirasaka, K.; Goto, J.; Nikawa, T.; Shono, M.; Yoshida, M.; Stoward, P.J. Subcutaneous injection, from birth, of epigallocatechin-3-gallate, a component of green tea, limits the onset of muscular dystrophy in mdx mice: A quantitative histological, immunohistochemical and electrophysiological study. Histochem Cell Biol. 2008, 129, 489–501. [Google Scholar] [CrossRef]
- Alway, S.E.; Bennett, B.T.; Wilson, J.C.; Sperringer, J.; Mohamed, J.S.; Edens, N.K.; Pereira, S.L. Green tea extract attenuates muscle loss and improves muscle function during disuse, but fails to improve muscle recovery following unloading in aged rats. J. Appl. Physiol. (1985) 2015, 118, 319–330. [Google Scholar] [CrossRef]
- Renno, W.M.; Benov, L.; Khan, K.M. Possible role of antioxidative capacity of (-)-epigallocatechin-3-gallate treatment in morphological and neurobehavioral recovery after sciatic nerve crush injury. J. Neurosurg. Spine 2017, 27, 593–613. [Google Scholar] [CrossRef] [PubMed]
- Alcazar, C.A.; Hu, C.; Rando, T.A.; Huang, N.F.; Nakayama, K.H. Transplantation of insulin-like growth factor-1 laden scaffolds combined with exercise promotes neuroregeneration and angiogenesis in a preclinical muscle injury model. Biomater. Sci. 2020, 8, 5376–5389. [Google Scholar] [CrossRef]
- Kang, S.H.; Lee, H.A.; Kim, M.; Lee, E.; Sohn, U.D.; Kim, I. Forkhead box O3 plays a role in skeletal muscle atrophy through expression of E3 ubiquitin ligases MuRF-1 and atrogin-1 in Cushing’s syndrome. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E495–E507. [Google Scholar] [CrossRef]
- Wimmer, R.J.; Russell, S.J.; Schneider, M.F. Green tea component EGCG, insulin and IGF-1 promote nuclear efflux of atrophy-associated transcription factor Foxo1 in skeletal muscle fibers. J. Nutr. Biochem. 2015, 26, 1559–1567. [Google Scholar] [CrossRef]
- Qian, Y.; Yao, Z.; Wang, X.; Cheng, Y.; Fang, Z.; Yuan, W.E.; Fan, C.; Ouyang, Y. (-)-Epigallocatechin gallate-loaded polycaprolactone scaffolds fabricated using a 3D integrated moulding method alleviate immune stress and induce neurogenesis. Cell Prolif. 2020, 53, e12730. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Lee, S.H.; Park, Y.; Lee, H.S.; Hong, J.S.; Lim, C.Y.; Kim, D.H.; Park, S.S.; Suh, H.J.; Hong, K.B. (-)-Epicatechin-Enriched Extract from Camellia sinensis Improves Regulation of Muscle Mass and Function: Results from a Randomized Controlled Trial. Antioxidants 2021, 10, 1026. [Google Scholar] [CrossRef] [PubMed]
- Mahler, A.; Steiniger, J.; Bock, M.; Klug, L.; Parreidt, N.; Lorenz, M.; Zimmermann, B.F.; Krannich, A.; Paul, F.; Boschmann, M. Metabolic response to epigallocatechin-3-gallate in relapsing-remitting multiple sclerosis: A randomized clinical trial. Am. J. Clin. Nutr. 2015, 101, 487–495. [Google Scholar] [CrossRef]
- Ulatowski, L.; Parker, R.; Warrier, G.; Sultana, R.; Butterfield, D.A.; Manor, D. Vitamin E is essential for Purkinje neuron integrity. Neuroscience 2014, 260, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Goudarzvand, M.; Javan, M.; Mirnajafi-Zadeh, J.; Mozafari, S.; Tiraihi, T. Vitamins E and D3 attenuate demyelination and potentiate remyelination processes of hippocampal formation of rats following local injection of ethidium bromide. Cell. Mol. Neurobiol. 2010, 30, 289–299. [Google Scholar] [CrossRef]
- Azizi, A.; Azizi, S.; Heshmatian, B.; Amini, K. Improvement of functional recovery of transected peripheral nerve by means of chitosan grafts filled with vitamin E, pyrroloquinoline quinone and their combination. Int. J. Surg. 2014, 12, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Glaser, V.; Moritz, B.; Schmitz, A.; Dafre, A.L.; Nazari, E.M.; Rauh Muller, Y.M.; Feksa, L.; Straliottoa, M.R.; de Bem, A.F.; Farina, M.; et al. Protective effects of diphenyl diselenide in a mouse model of brain toxicity. Chem. Biol. Interact. 2013, 206, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Moghadaszadeh, B.; Beggs, A.H. Selenoproteins and their impact on human health through diverse physiological pathways. Physiology (Bethesda) 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Solovyev, N.D. Importance of selenium and selenoprotein for brain function: From antioxidant protection to neuronal signalling. J. Inorg. Biochem. 2015, 153, 1–12. [Google Scholar] [CrossRef]
- Kiyohara, A.C.P.; Torres, D.J.; Hagiwara, A.; Pak, J.; Rueli, R.; Shuttleworth, C.W.R.; Bellinger, F.P. Selenoprotein P Regulates Synaptic Zinc and Reduces Tau Phosphorylation. Front. Nutr. 2021, 8, 683154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Cai, W.; Jin, M.; Xu, J.; Wang, Y.; Xiao, Y.; Hao, L.; Wang, B.; Zhang, Y.; Han, J.; et al. 18beta-glycyrrhetinic acid suppresses experimental autoimmune encephalomyelitis through inhibition of microglia activation and promotion of remyelination. Sci. Rep. 2015, 5, 13713. [Google Scholar] [CrossRef] [PubMed]
- Lozinski, B.M.; de Almeida, L.G.N.; Silva, C.; Dong, Y.; Brown, D.; Chopra, S.; Yong, V.W.; Dufour, A. Exercise rapidly alters proteomes in mice following spinal cord demyelination. Sci. Rep. 2021, 11, 7239. [Google Scholar] [CrossRef]
- Saito, Y.; Chikenji, T.S.; Matsumura, T.; Nakano, M.; Fujimiya, M. Exercise enhances skeletal muscle regeneration by promoting senescence in fibro-adipogenic progenitors. Nat. Commun. 2020, 11, 889. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.K.; Michaels, N.J.; Ilyntskyy, S.; Keough, M.B.; Kovalchuk, O.; Yong, V.W. Multimodal Enhancement of Remyelination by Exercise with a Pivotal Role for Oligodendroglial PGC1alpha. Cell Rep. 2018, 24, 3167–3179. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.C.; Reynolds, E.; Banerjee, M.; Chant, E.; Villegas-Umana, E.; Feldman, E.L. Central Obesity is Associated with Neuropathy in the Severely Obese. Mayo Clin. Proc. 2020, 95, 1342–1353. [Google Scholar] [CrossRef]
- Bekar, E.; Altunkaynak, B.Z.; Balci, K.; Aslan, G.; Ayyildiz, M.; Kaplan, S. Effects of high fat diet induced obesity on peripheral nerve regeneration and levels of GAP 43 and TGF-beta in rats. Biotech. Histochem. 2014, 89, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Peng, J.; Han, G.H.; Ding, X.; Wei, S.; Gao, G.; Huang, K.; Chang, F.; Wang, Y. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 2019, 14, 1335–1342. [Google Scholar] [CrossRef]
- Iqbal, Z.; Bashir, B.; Ferdousi, M.; Kalteniece, A.; Alam, U.; Malik, R.A.; Soran, H. Lipids and peripheral neuropathy. Curr. Opin Lipidol. 2021, 32, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, M.; Zhang, S.; Foretz, M.; Viollet, B.; Du, M. Obesity Impairs Skeletal Muscle Regeneration Through Inhibition of AMPK. Diabetes 2016, 65, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torres, I.; Castrejon-Tellez, V.; Soto, M.E.; Rubio-Ruiz, M.E.; Manzano-Pech, L.; Guarner-Lans, V. Oxidative Stress, Plant Natural Antioxidants, and Obesity. Int. J. Mol. Sci. 2021, 22, 1786. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabezas Perez, R.J.; Ávila Rodríguez, M.F.; Rosero Salazar, D.H. Exogenous Antioxidants in Remyelination and Skeletal Muscle Recovery. Biomedicines 2022, 10, 2557. https://doi.org/10.3390/biomedicines10102557
Cabezas Perez RJ, Ávila Rodríguez MF, Rosero Salazar DH. Exogenous Antioxidants in Remyelination and Skeletal Muscle Recovery. Biomedicines. 2022; 10(10):2557. https://doi.org/10.3390/biomedicines10102557
Chicago/Turabian StyleCabezas Perez, Ricardo Julián, Marco Fidel Ávila Rodríguez, and Doris Haydee Rosero Salazar. 2022. "Exogenous Antioxidants in Remyelination and Skeletal Muscle Recovery" Biomedicines 10, no. 10: 2557. https://doi.org/10.3390/biomedicines10102557
APA StyleCabezas Perez, R. J., Ávila Rodríguez, M. F., & Rosero Salazar, D. H. (2022). Exogenous Antioxidants in Remyelination and Skeletal Muscle Recovery. Biomedicines, 10(10), 2557. https://doi.org/10.3390/biomedicines10102557