

Association of Circular RNA and Long Non-Coding RNA Dysregulation with the Clinical Response to Immune Checkpoint Blockade in Cutaneous Metastatic Melanoma
 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Nucleic Acid Isolation
2.3. Next Generation Sequencing
2.4. Realtime PCR Validation
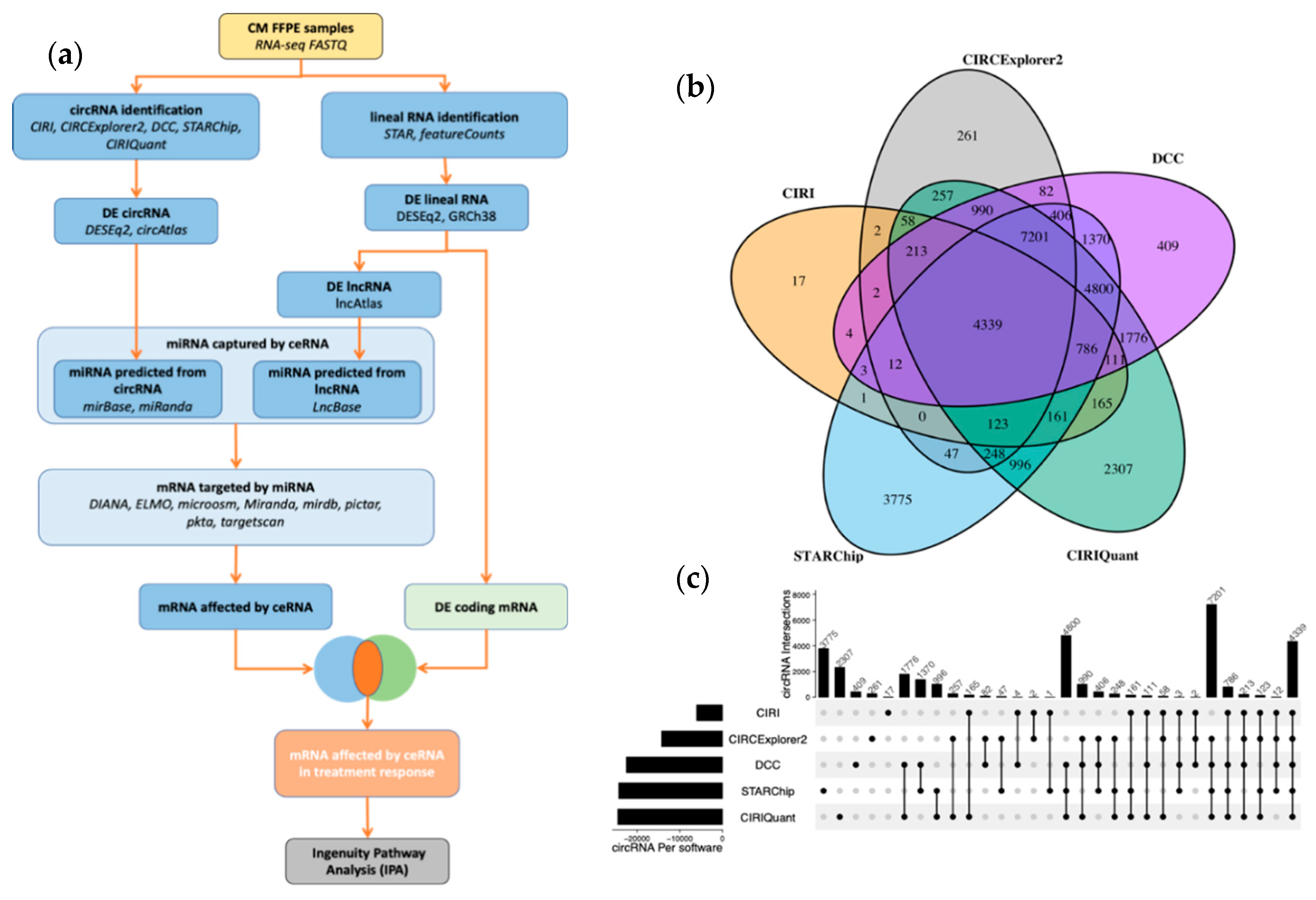
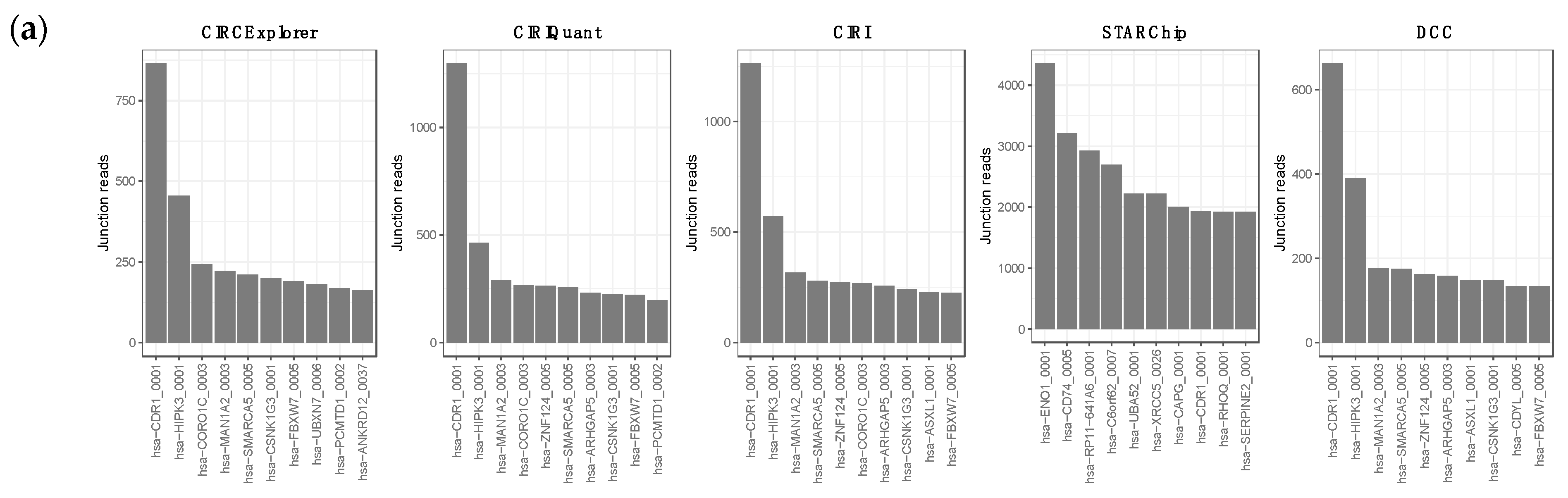
2.5. lncRNA and circRNA Detection
2.6. Differential Expression Analysis
2.7. ceRNAs-miRNAs-mRNAs Interactions
2.8. Gene Set Enrichment and Gene Interactions Networks
2.9. Statistics and Visualization
2.10. Special Case
3. Results
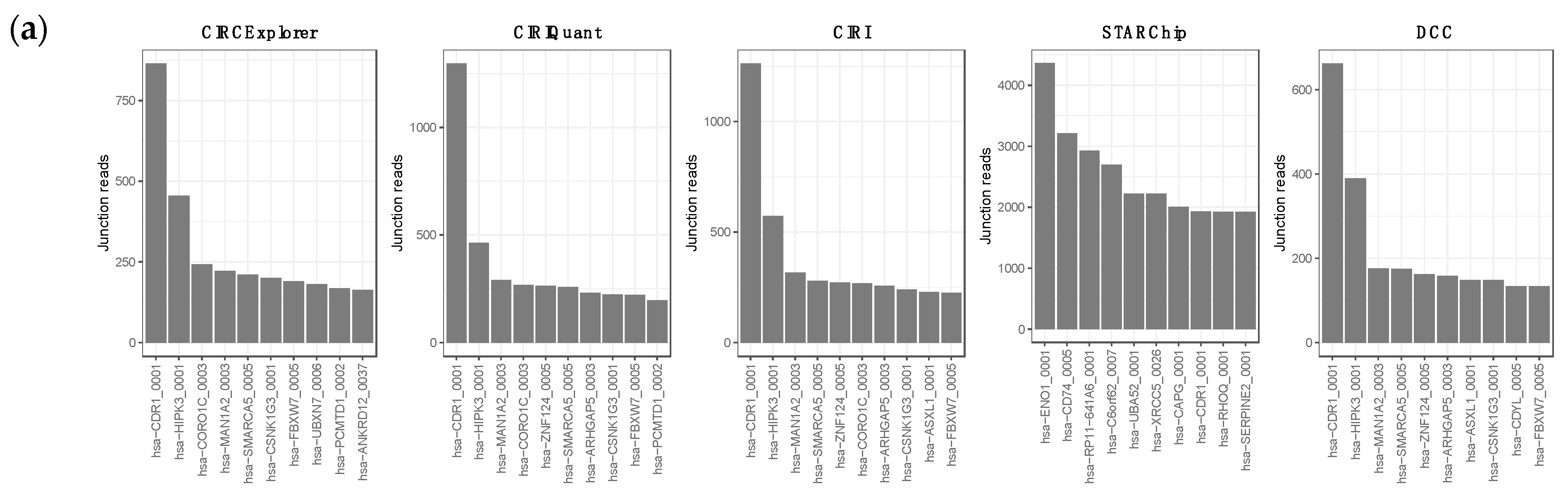
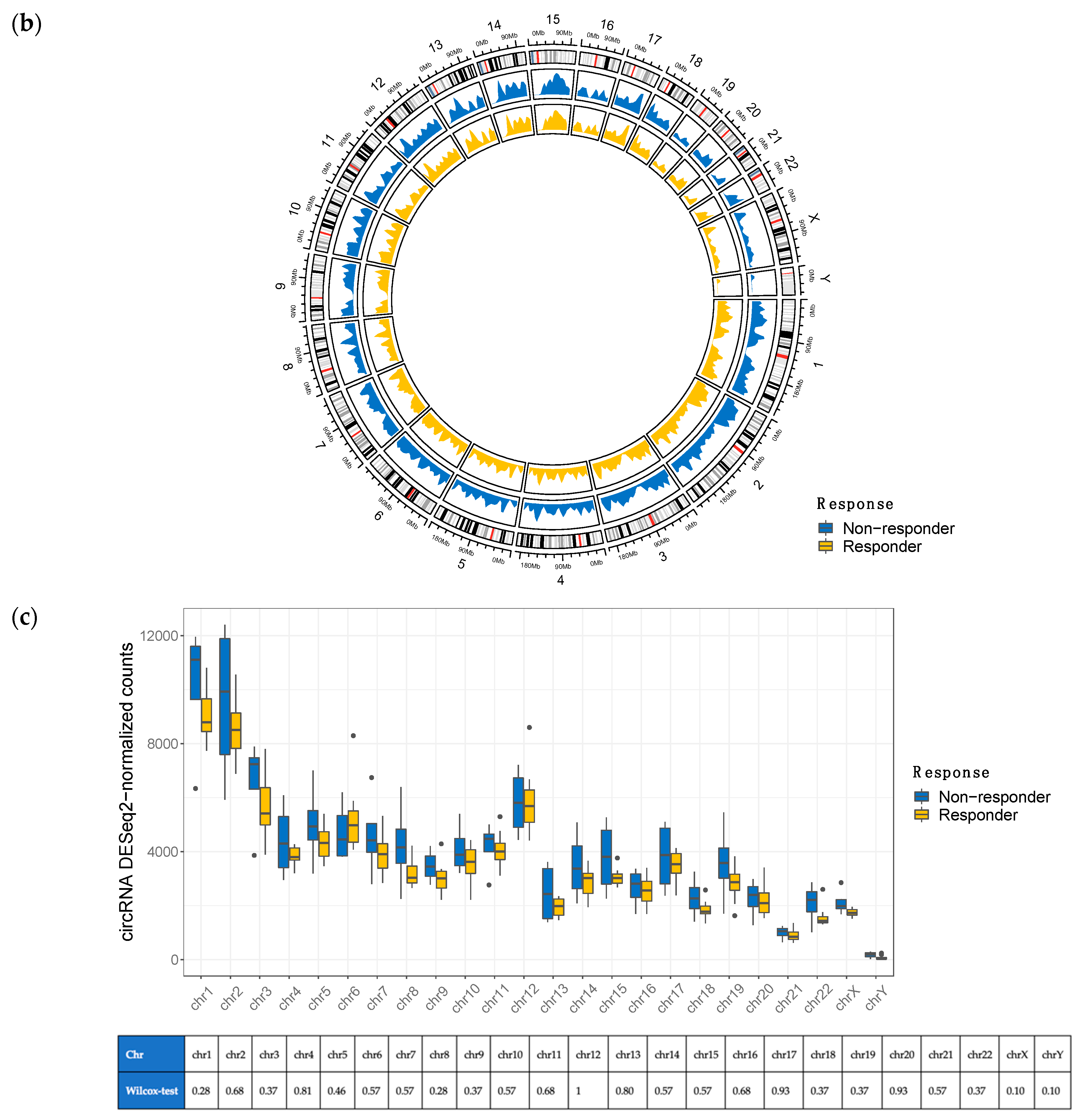
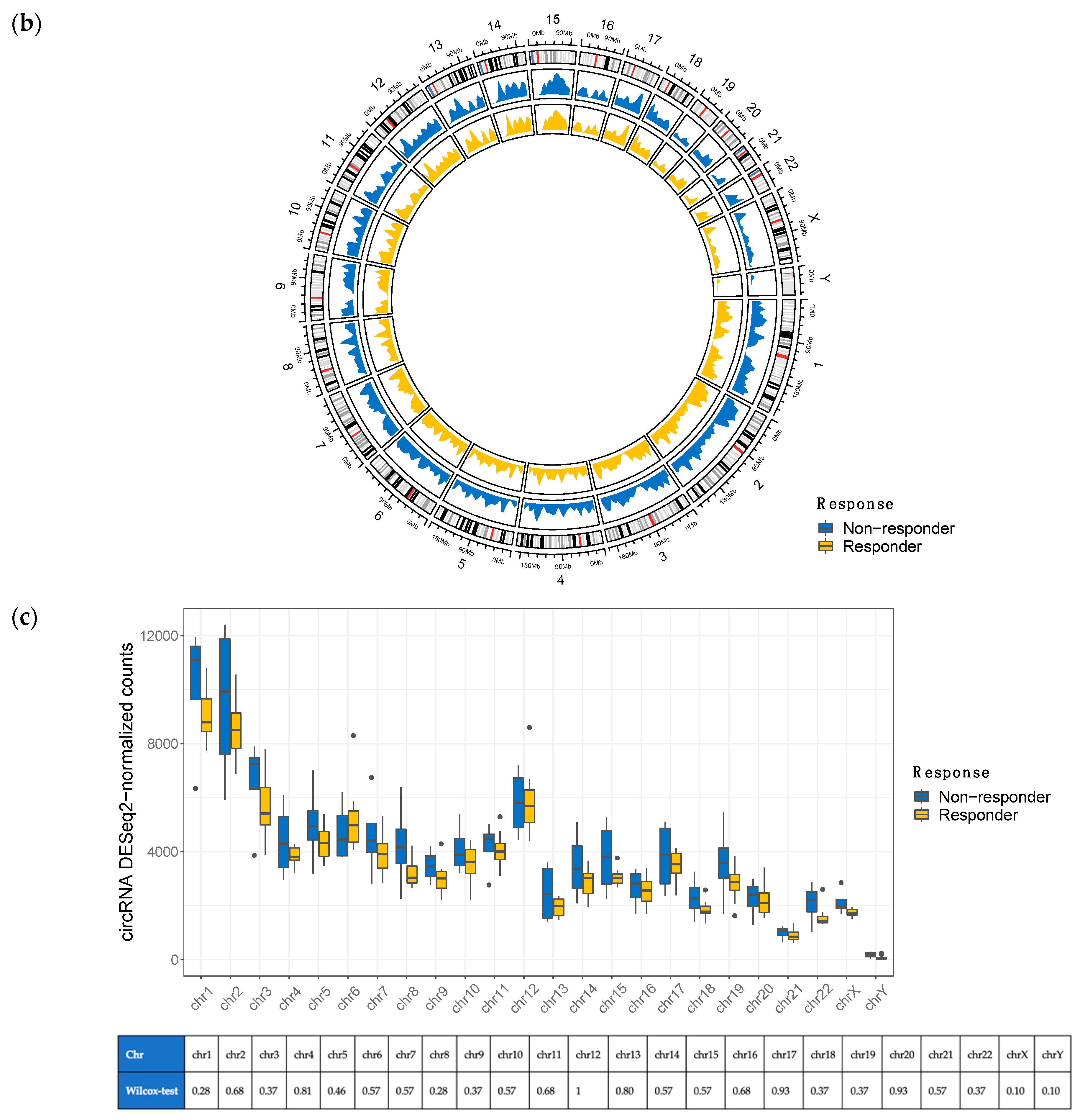
3.1. Overview of circRNA and lncRNA Expression Patterns in Cutaneous Melanoma Tissues
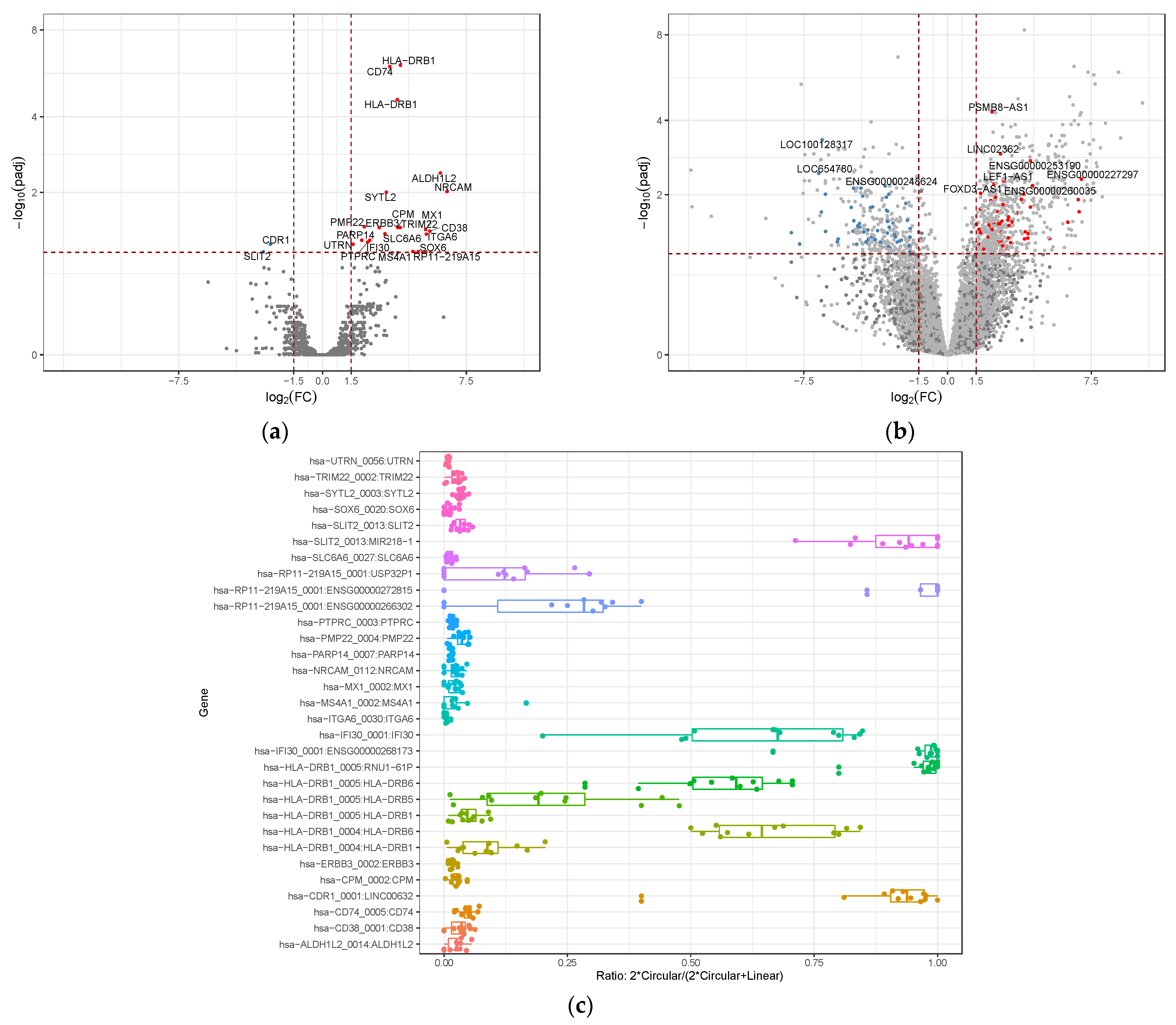
3.2. Differential Gene Expression of circRNAs and lncRNAs
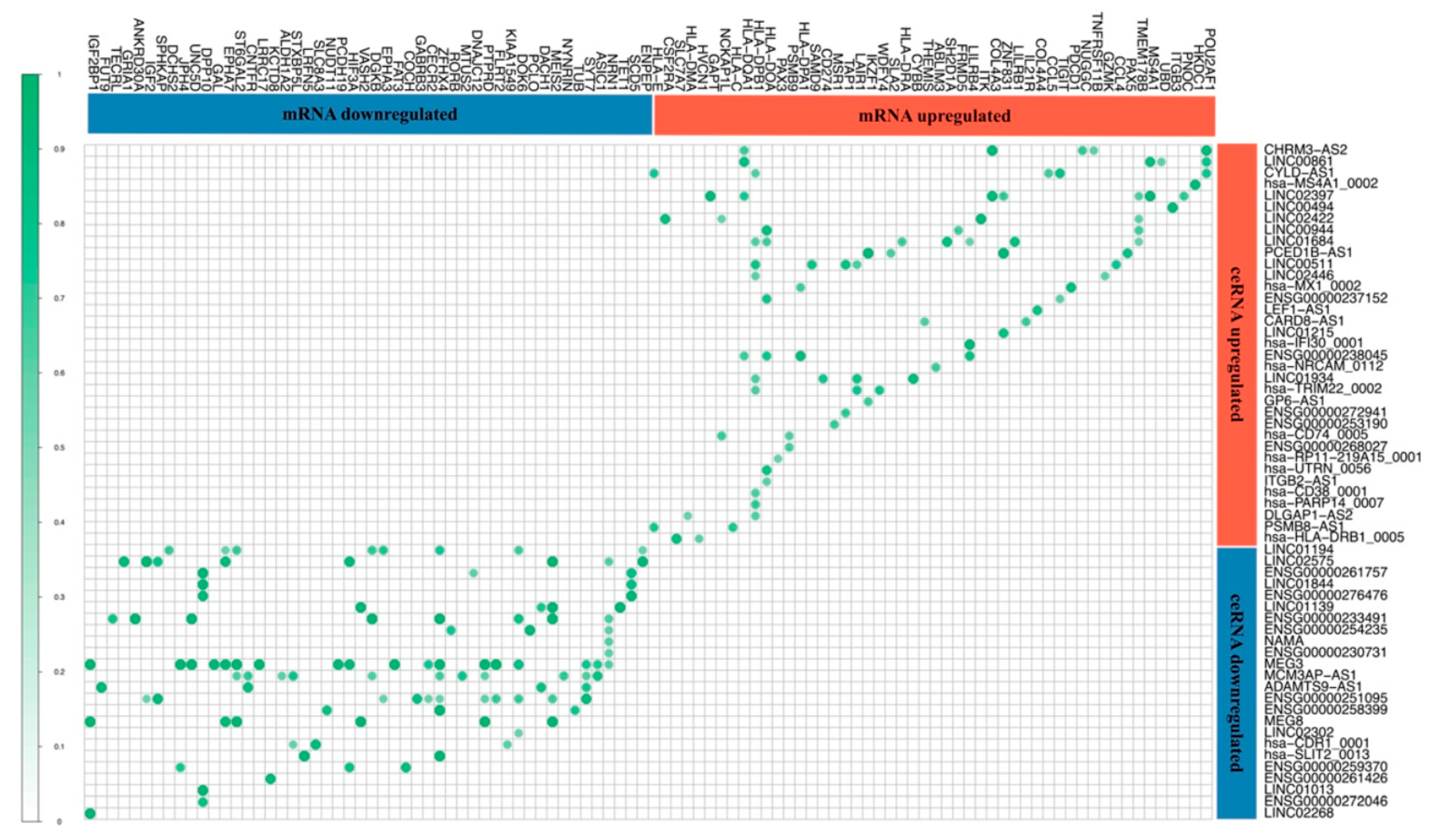
3.3. Competitor Endogenous RNA Network (ceRNA Network)
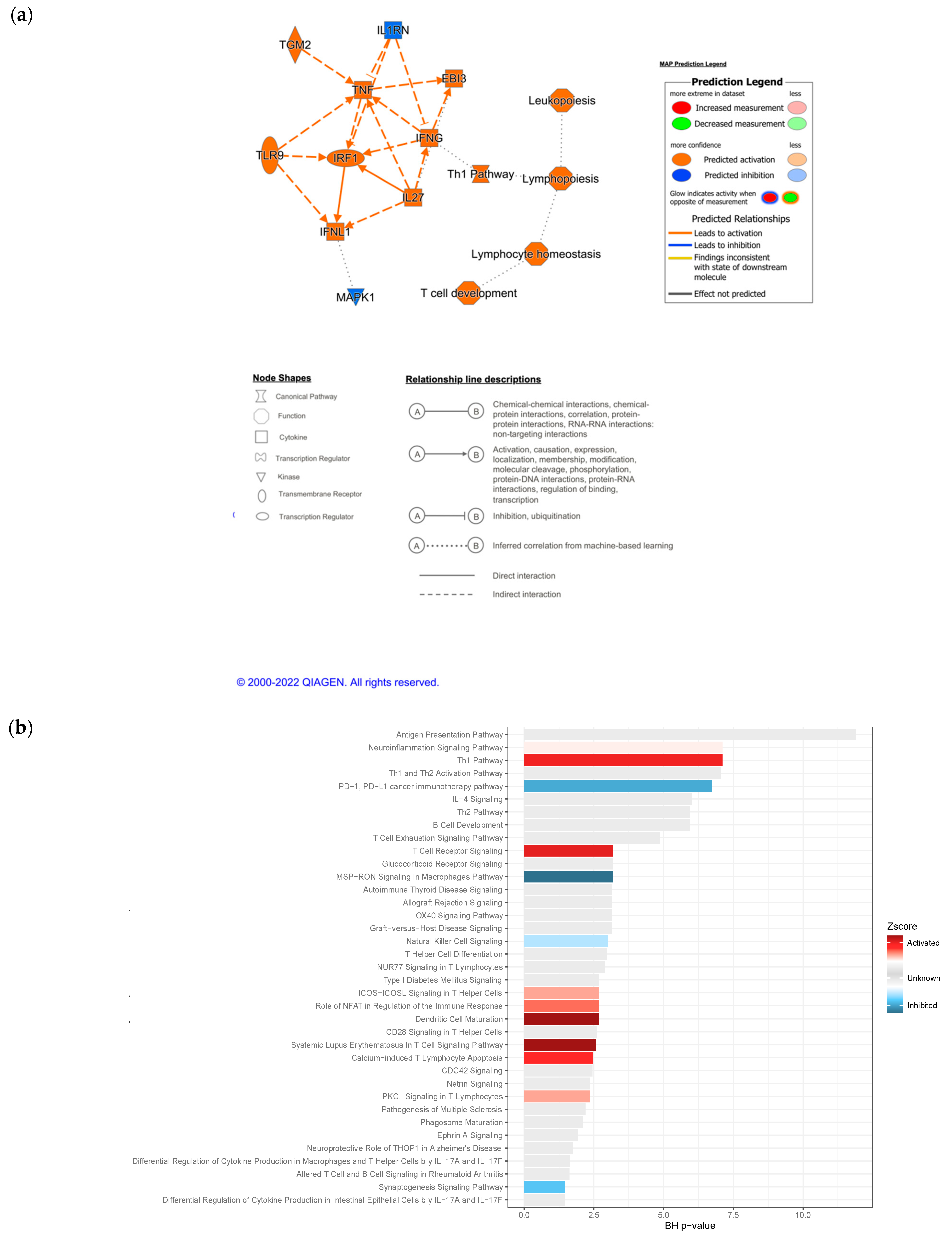
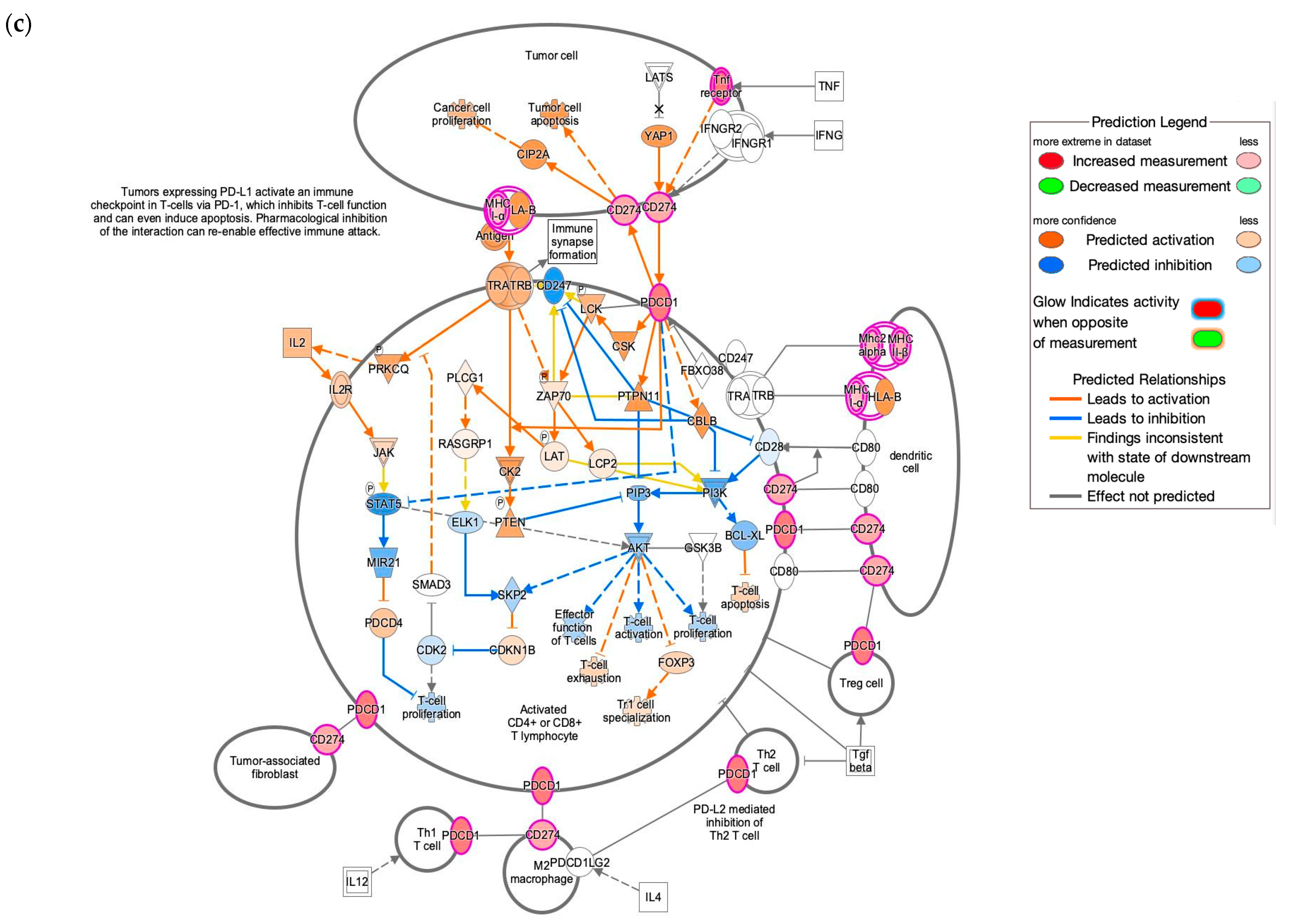
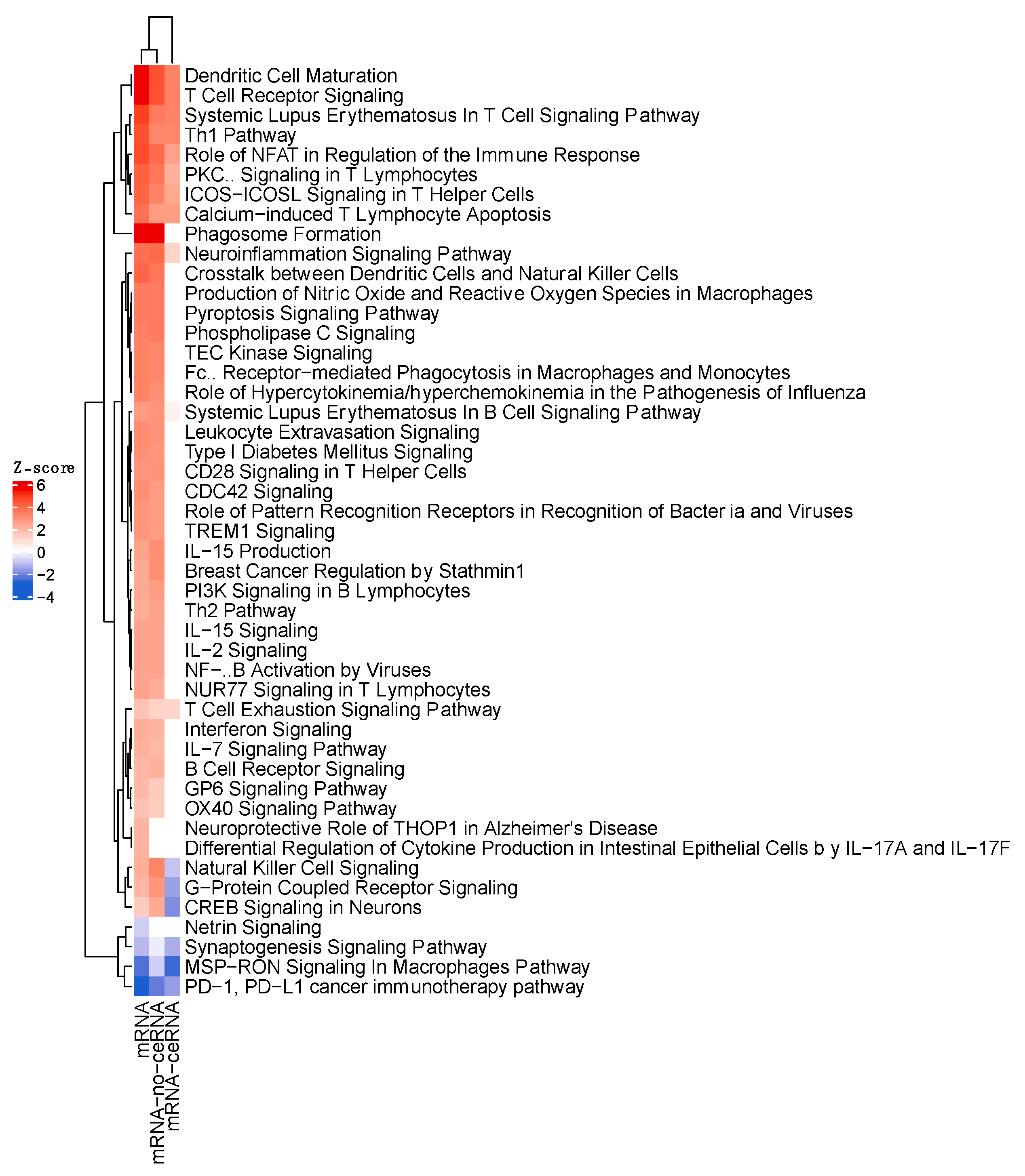
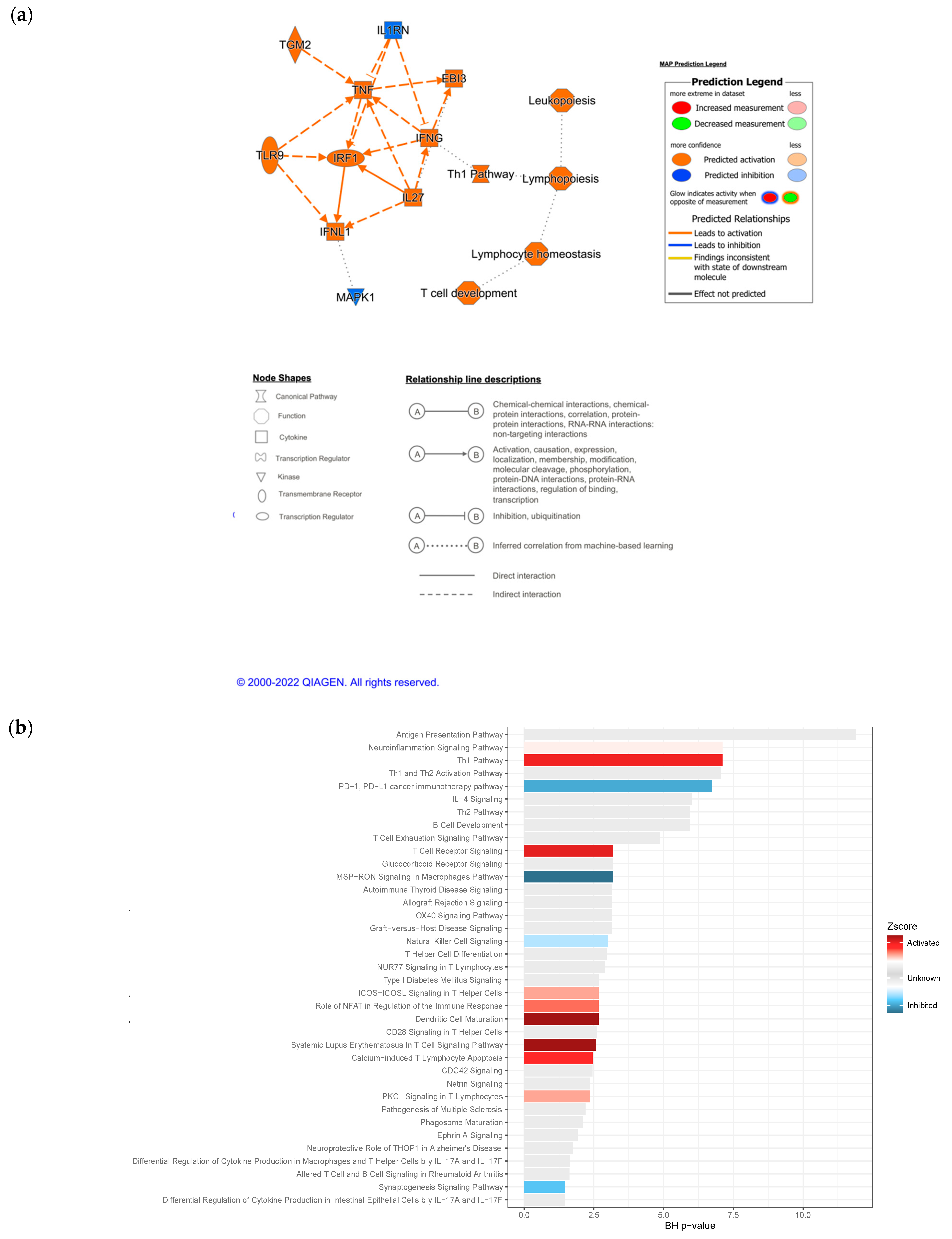
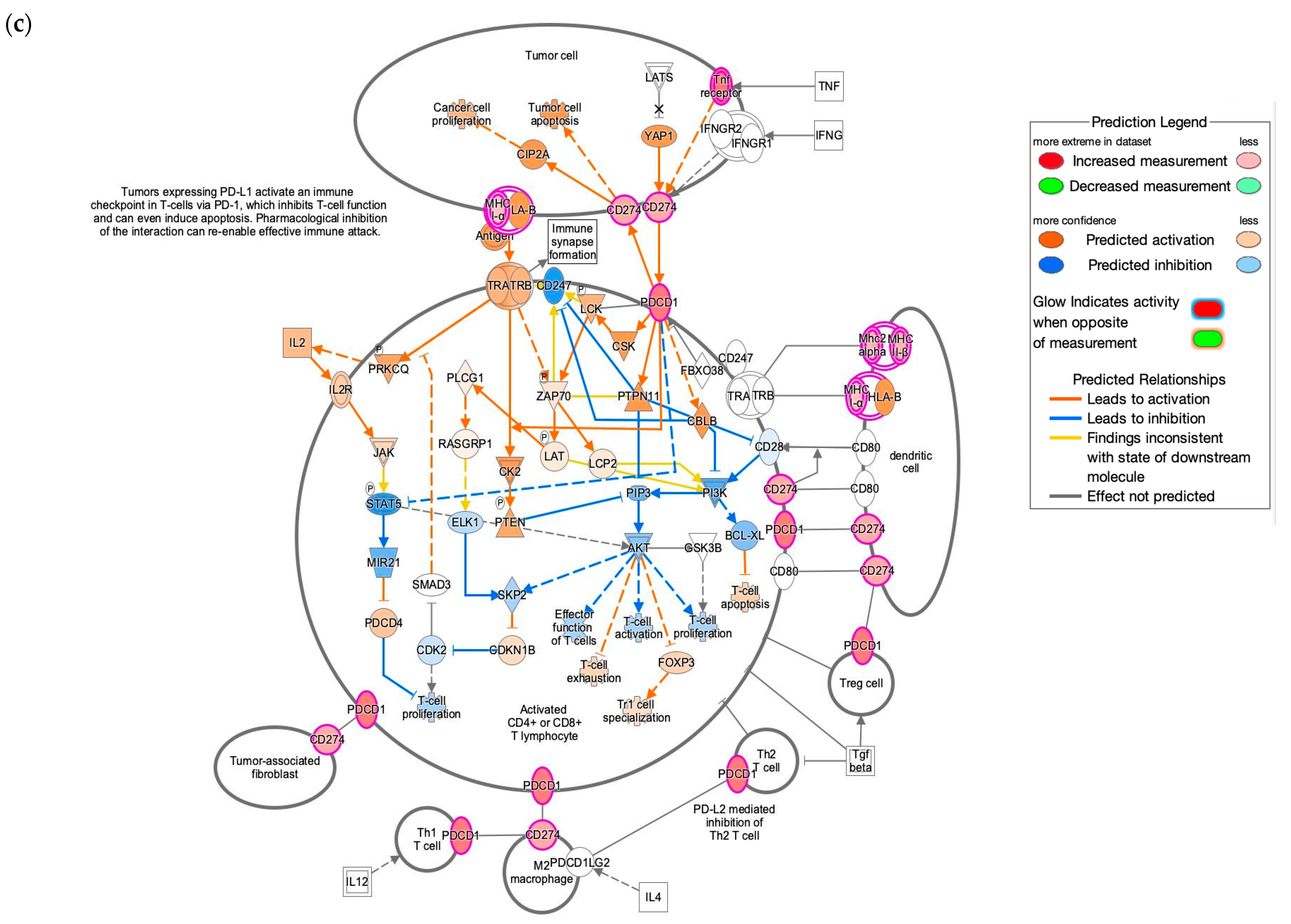
3.4. IPA Functional Enrichment Analysis Based on the ceRNA Network
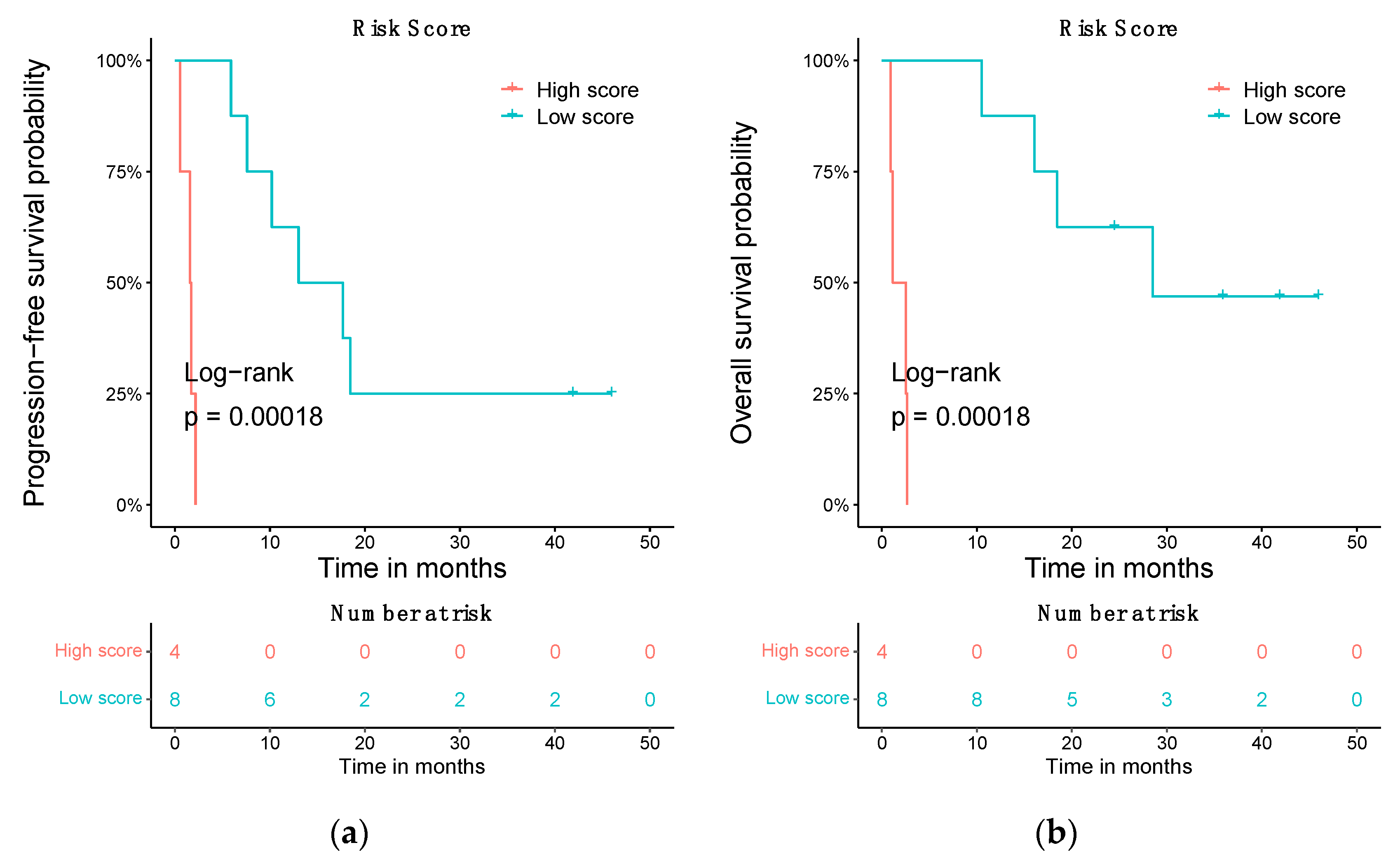
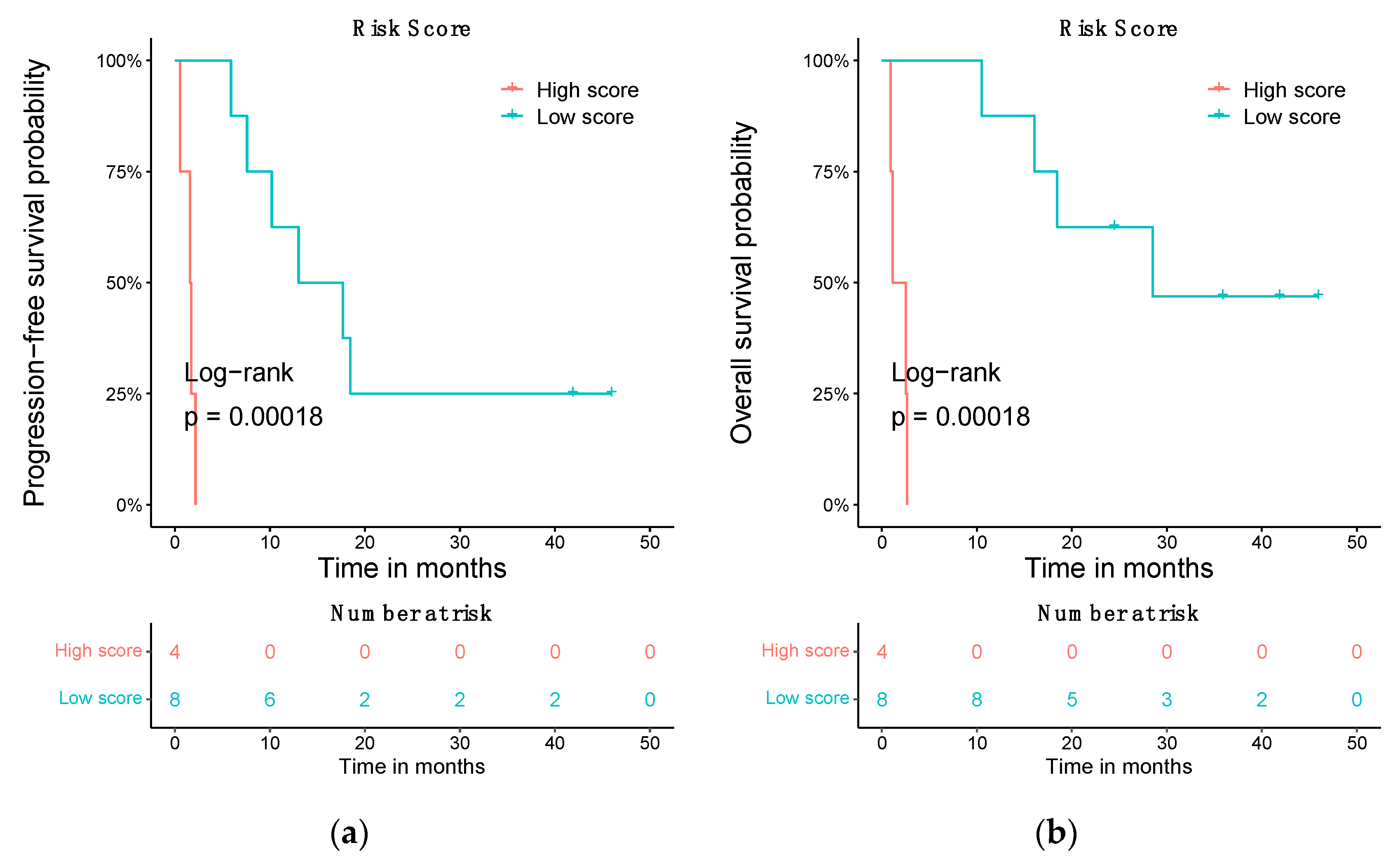
3.5. Prognostic Risk Score Using the Differentially Expressed ceRNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shakhova, O. Neural Crest Stem Cells in Melanoma Development. Curr. Opin. Oncol. 2014, 26, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Fukunaga-Kalabis, M.; Herlyn, M. Crosstalk in Skin: Melanocytes, Keratinocytes, Stem Cells, and Melanoma. J. Cell Commun. Signal. 2016, 10, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Erdei, E.; Torres, S.M. A New Understanding in the Epidemiology of Melanoma. Expert Rev. Anticancer Ther. 2010, 10, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Laversanne, M.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.B.F. Global Cancer Observatory: Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow (accessed on 9 March 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.C.; Seebode, C.; Lehmann, J.; Emmert, S. Photocarcinogenesis and Skin Cancer Prevention Strategies: An Update. Anticancer Res. 2018, 38, 1153–1158. [Google Scholar] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Palmieri, G.; Ombra, M.; Colombino, M.; Casula, M.; Sini, M.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 1. [Google Scholar] [CrossRef]
- Damsky, W.E.; Bosenberg, M. Melanocytic Nevi and Melanoma: Unraveling a Complex Relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef]
- Ding, L.; Kim, M.; Kanchi, K.L.; Dees, N.D.; Lu, C.; Griffith, M.; Fenstermacher, D.; Sung, H.; Miller, C.A.; Goetz, B.; et al. Clonal Architectures and Driver Mutations in Metastatic Melanomas. PLoS ONE 2014, 9, e111153. [Google Scholar] [CrossRef]
- Vergara, I.A.; Mintoff, C.P.; Sandhu, S.; McIntosh, L.; Young, R.J.; Wong, S.Q.; Colebatch, A.; Cameron, D.L.; Kwon, J.L.; Wolfe, R.; et al. Evolution of Late-Stage Metastatic Melanoma Is Dominated by Aneuploidy and Whole Genome Doubling. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.-C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.v. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Filipovic, A.; Miller, G.; Bolen, J. Progress Toward Identifying Exact Proxies for Predicting Response to Immunotherapies. Front. Cell Dev. Biol. 2020, 8, 155. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The Multilayered Complexity of CeRNA Crosstalk and Competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The Landscape of Long Noncoding RNAs in the Human Transcriptome. Nat. Genet. 2015, 47, 199. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The Biogenesis, Biology and Characterization of Circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Montico, B.; Giurato, G.; Pecoraro, G.; Salvati, A.; Covre, A.; Colizzi, F.; Steffan, A.; Weisz, A.; Maio, M.; Sigalotti, L.; et al. The Pleiotropic Role of Circular and Long Noncoding RNAs in Cutaneous Melanoma. Mol. Oncol. 2021, 16, 565–593. [Google Scholar] [CrossRef]
- Jiang, W.; Pan, S.; Chen, X.; Wang, Z.; Zhu, X. The Role of LncRNAs and CircRNAs in the PD-1/PD-L1 Pathway in Cancer Immunotherapy. Mol. Cancer 2021, 20, 1–17. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient MicroRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ding, R.F.; Sun, Y.C.; Huo, S.T.; He, A.; Wen, C.; Chen, H.H.; Du, W.W.; Lai, W.N.; Wang, H. Circular RNA in Tumor Metastasis. Mol. Ther. Nucleic Acids 2021, 23, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An Efficient and Unbiased Algorithm for de Novo Circular RNA Identification. Genome Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Dong, R.; Zhang, Y.; Zhang, J.L.; Luo, Z.; Zhang, J.; Chen, L.L.; Yang, L. Diverse Alternative Back-Splicing and Alternative Splicing Landscape of Circular RNAs. Genome Res. 2016, 26, 1277–1287. [Google Scholar] [CrossRef]
- Cheng, J.; Metge, F.; Dieterich, C. Specific Identification and Quantification of Circular RNAs from Sequencing Data. Bioinformatics 2016, 32, 1094–1096. [Google Scholar] [CrossRef]
- Akers, N.K.; Schadt, E.E.; Losic, B. STAR Chimeric Post for Rapid Detection of Circular RNA and Fusion Transcripts. Bioinformatics 2018, 34, 2364–2370. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Yang, J.; Zhao, F. Accurate Quantification of Circular RNAs Identifies Extensive Circular Isoform Switching Events. Nat. Commun. 2020, 11, 90. [Google Scholar] [CrossRef]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An Integrated Resource of One Million Highly Accurate Circular RNAs from 1070 Vertebrate Transcriptomes. Genome Biol. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Pulido, T.H.; Guigo, R.; Johnson, R. LncATLAS Database for Subcellular Localization of Long Noncoding RNAs. Rna 2017, 23, 1080–1087. [Google Scholar] [CrossRef]
- Yao, D.; Zhang, L.; Zheng, M.; Sun, X.; Lu, Y.; Liu, P. Circ2Disease: A Manually Curated Database of Experimentally Validated CircRNAs in Human Disease. Sci. Rep. 2018, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Rophina, M.; Sharma, D.; Poojary, M.; Scaria, V. Circad: A Comprehensive Manually Curated Resource of Circular RNA Associated with Diseases. Database 2020, 2020, 19. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Feng, J.; Chen, K.; Ma, Y.; Gong, J.; Cai, F.; Jin, Y.; Gao, Y.; Xia, L.; Chang, H.; et al. CSCD: A Database for Cancer-Specific Circular RNAs. Nucleic Acids Res. 2018, 46, D925–D929. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lee, Y.C.; Chang, K.L.; Hsiao, K.Y. Analysis of Common Targets for Circular RNAs. BMC Bioinformatics 2019, 20, 372. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA Targets in Drosophila. Genome Biol. 2003, 5, 1–14. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Vlachos, I.S.; Karagkouni, D.; Georgakilas, G.; Kanellos, I.; Vergoulis, T.; Zagganas, K.; Tsanakas, P.; Floros, E.; Dalamagas, T.; et al. DIANA-LncBase v2: Indexing MicroRNA Targets on Non-Coding Transcripts. Nucleic Acids Res. 2016, 44, D231–D238. [Google Scholar] [CrossRef]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The MultiMiR R Package and Database: Integration of MicroRNA-Target Interactions along with Their Disease and Drug Associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Maragkakis, M.; Vergoulis, T.; Alexiou, P.; Reczko, M.; Plomaritou, K.; Gousis, M.; Kourtis, K.; Koziris, N.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-MicroT Web Server Upgrade Supports Fly and Worm MiRNA Target Prediction and Bibliographic MiRNA to Disease Association. Nucleic Acids Res. 2011, 39, W145. [Google Scholar] [CrossRef]
- Gaidatzis, D.; van Nimwegen, E.; Hausser, J.; Zavolan, M. Inference of MiRNA Targets Using Evolutionary Conservation and Pathway Analysis. BMC Bioinformatics 2007, 8, 69. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. MiRBase: Tools for MicroRNA Genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [Green Version]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The MicroRNA.Org Resource: Targets and Expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. MiRDB: A MicroRNA Target Prediction and Functional Annotation Database with a Wiki Interface. RNA 2008, 14, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Dieterich, C.; Wurmus, R.; Rajewsky, N.; Landthaler, M.; Akalin, A. DoRiNA 2.0-Upgrading the Dorina Database of RNA Interactions in Post-Transcriptional Regulation. Nucleic Acids Res. 2015, 43, D160–D167. [Google Scholar] [CrossRef]
- Akbari Moqadam, F.; Pieters, R.; Den Boer, M.L. The Hunting of Targets: Challenge in MiRNA Research. Leukemia 2013, 27, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- QIAGEN Inc. Ingenuity Pathway Analysis (IPA). Available online: https://www.qiagen.com/us/products/discovery-and-translational-research/next-generation-sequencing/informatics-and-data/interpretation-content-databases/ingenuity-pathway-analysis/ (accessed on 30 September 2021).
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2; Springer: New York, NY, USA, 2009. [Google Scholar]
- Wang, W.; Zhu, D.; Zhao, Z.; Sun, M.; Wang, F.; Li, W.; Zhang, J.; Jiang, G. RNA Sequencing Reveals the Expression Profiles of CircRNA and Identifies a Four-CircRNA Signature Acts as a Prognostic Marker in Esophageal Squamous Cell Carcinoma. Cancer Cell Int. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Onieva, J.L.; Xiao, Q.; Berciano-Guerrero, M.-Á.; Laborda-Illanes, A.; de Andrea, C.; Chaves, P.; Piñeiro, P.; Garrido-Aranda, A.; Gallego, E.; Sojo, B.; et al. High IGKC-Expressing Intratumoral Plasma Cells Predict Response to Immune Checkpoint Blockade. Int. J. Mol. Sci. 2022, 23, 9124. [Google Scholar] [CrossRef]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the Crosstalk between Melanoma and Immune Cells in the Tumor Microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.-H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Article 2021, 11, 1. [Google Scholar] [CrossRef]
- Carlos-Reyes, Á.; Romero-Garcia, S.; Contreras-Sanzón, E.; Ruiz, V.; Prado-Garcia, H. Role of Circular RNAs in the Regulation of Immune Cells in Response to Cancer Therapies. Front. Genet. 2022, 13, 823238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dang, D.; Cong, L.; Sun, H.; Cong, X. Pivotal Factors Associated with the Immunosuppressive Tumor Microenvironment and Melanoma Metastasis. Cancer Med. 2021, 10, 4710–4720. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Krasagakis, K. Effects of Interferons and Cytokines on Melanoma Cells. J. Investig. Dermatol. 1993, 100, S239–S244. [Google Scholar] [CrossRef]
- Weigelin, B.; den Boer, A.T.; Wagena, E.; Broen, K.; Dolstra, H.; de Boer, R.J.; Figdor, C.G.; Textor, J.; Friedl, P. Cytotoxic T Cells Are Able to Efficiently Eliminate Cancer Cells by Additive Cytotoxicity. Nat. Commun. 2021, 12, 5217. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα Blockade Overcomes Resistance to Anti-PD-1 in Experimental Melanoma. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hanniford, D.; Ulloa-Morales, A.; Karz, A.; Berzoti-Coelho, M.G.; Moubarak, R.S.; Sánchez-Sendra, B.; Kloetgen, A.; Davalos, V.; Imig, J.; Wu, P.; et al. Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell 2020, 37, 55–70.e15. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliver, J.; Onieva, J.L.; Garrido-Barros, M.; Berciano-Guerrero, M.-Á.; Sánchez-Muñoz, A.; José Lozano, M.; Farngren, A.; Álvarez, M.; Martínez-Gálvez, B.; Pérez-Ruiz, E.; et al. Association of Circular RNA and Long Non-Coding RNA Dysregulation with the Clinical Response to Immune Checkpoint Blockade in Cutaneous Metastatic Melanoma. Biomedicines 2022, 10, 2419. https://doi.org/10.3390/biomedicines10102419
Oliver J, Onieva JL, Garrido-Barros M, Berciano-Guerrero M-Á, Sánchez-Muñoz A, José Lozano M, Farngren A, Álvarez M, Martínez-Gálvez B, Pérez-Ruiz E, et al. Association of Circular RNA and Long Non-Coding RNA Dysregulation with the Clinical Response to Immune Checkpoint Blockade in Cutaneous Metastatic Melanoma. Biomedicines. 2022; 10(10):2419. https://doi.org/10.3390/biomedicines10102419
Chicago/Turabian StyleOliver, Javier, Juan Luis Onieva, Maria Garrido-Barros, Miguel-Ángel Berciano-Guerrero, Alfonso Sánchez-Muñoz, María José Lozano, Angela Farngren, Martina Álvarez, Beatriz Martínez-Gálvez, Elisabeth Pérez-Ruiz, and et al. 2022. "Association of Circular RNA and Long Non-Coding RNA Dysregulation with the Clinical Response to Immune Checkpoint Blockade in Cutaneous Metastatic Melanoma" Biomedicines 10, no. 10: 2419. https://doi.org/10.3390/biomedicines10102419
APA StyleOliver, J., Onieva, J. L., Garrido-Barros, M., Berciano-Guerrero, M.-Á., Sánchez-Muñoz, A., José Lozano, M., Farngren, A., Álvarez, M., Martínez-Gálvez, B., Pérez-Ruiz, E., Alba, E., Cobo, M., Rueda-Domínguez, A., & Barragán, I. (2022). Association of Circular RNA and Long Non-Coding RNA Dysregulation with the Clinical Response to Immune Checkpoint Blockade in Cutaneous Metastatic Melanoma. Biomedicines, 10(10), 2419. https://doi.org/10.3390/biomedicines10102419