

Next-Generation SINE Compound KPT−8602 Ameliorates Dystrophic Pathology in Zebrafish and Mouse Models of DMD
 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Zebrafish Care and KPT−8602 Dosing Experiments
2.2. Mice
2.3. KPT−8602 Drug Treatment in Mice
2.4. Open Field Test–Basal Activity Tracking
2.5. Histological Analysis
2.6. Muscle Single-Cell Suspension
2.7. Flow Cytometry Analysis
2.8. Osteopontin ELISA
2.9. Statistical Analysis
3. Results
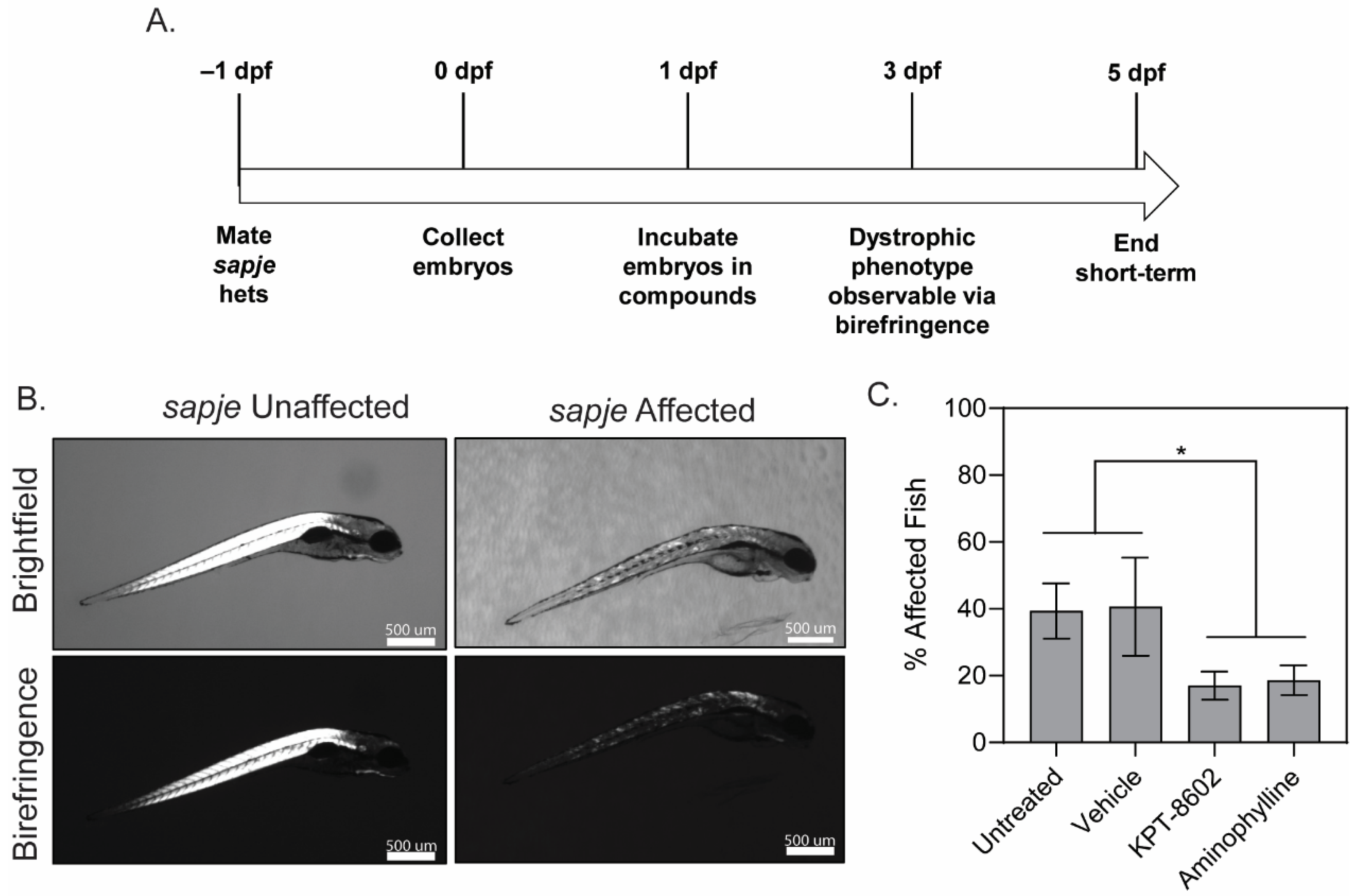
3.1. KPT−8602 Treatment Reduced Dystrophic Muscle Pathology in Zebrafish Model of DMD
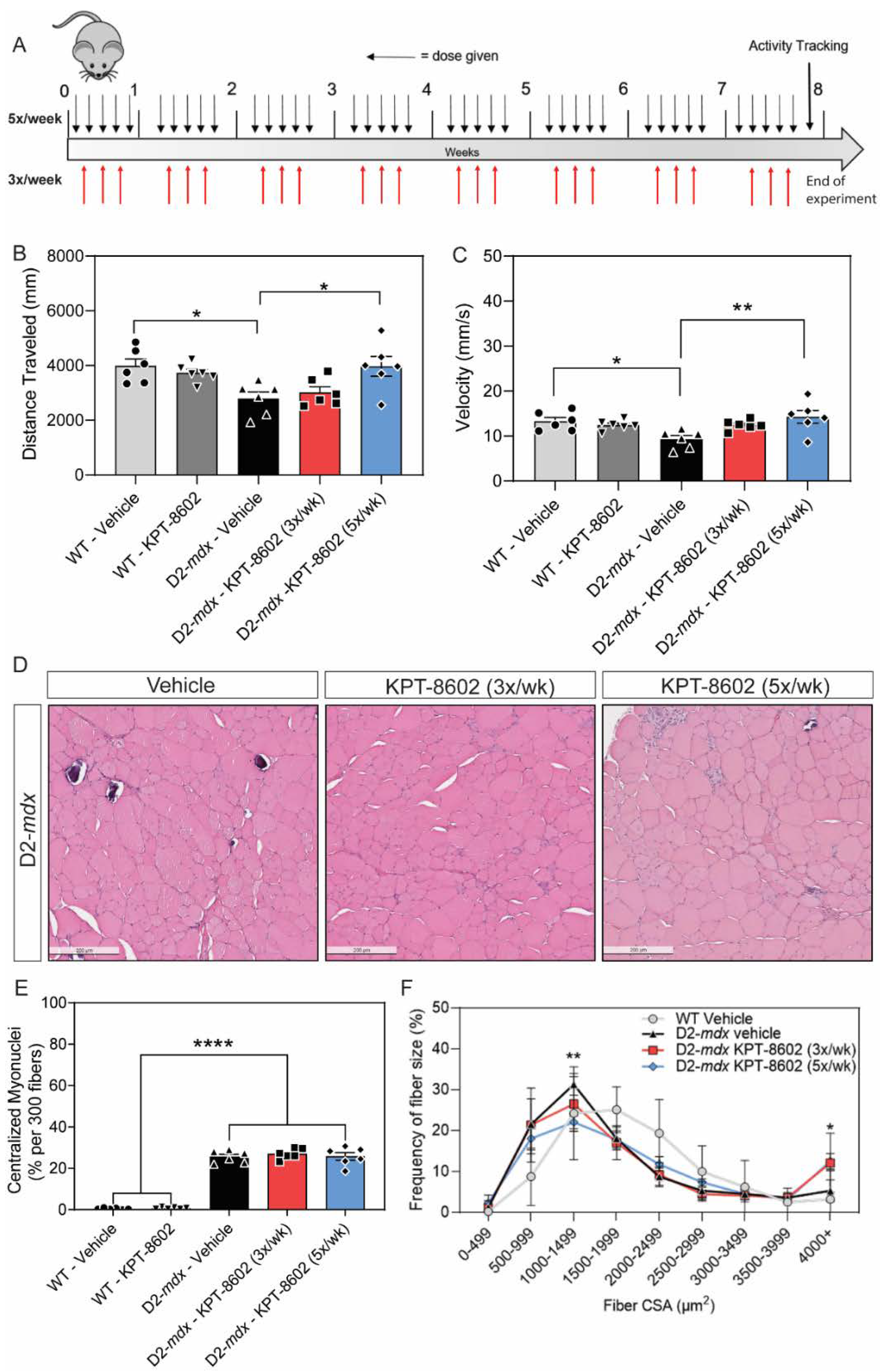
3.2. D2-Mdx Mice Treated with 5x/Week KPT−8602 Had Significantly Improved Activity and Myofiber Size
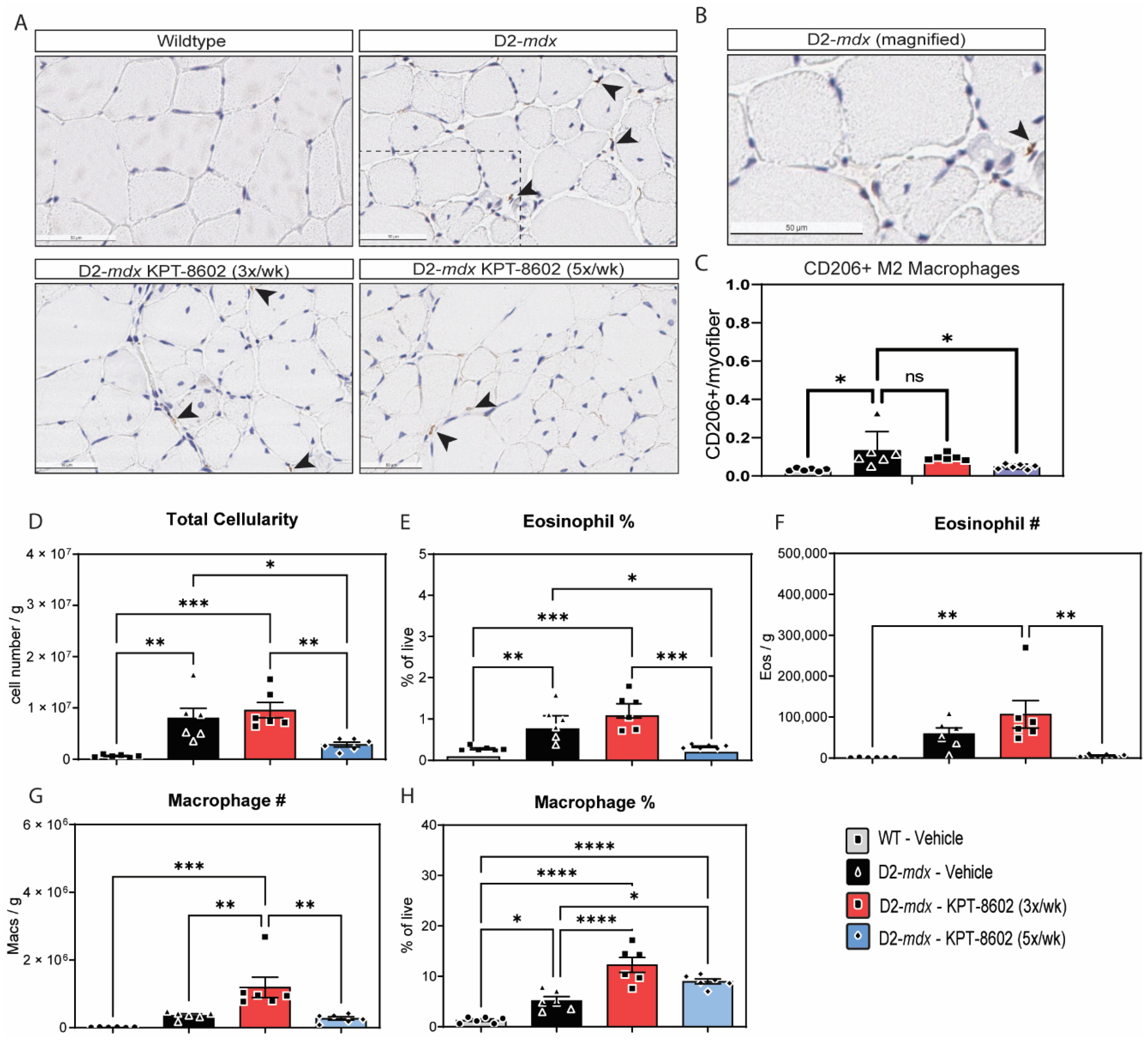
3.3. KPT−8602 Improves Immunological Profiles in Dystrophic Mouse Muscles
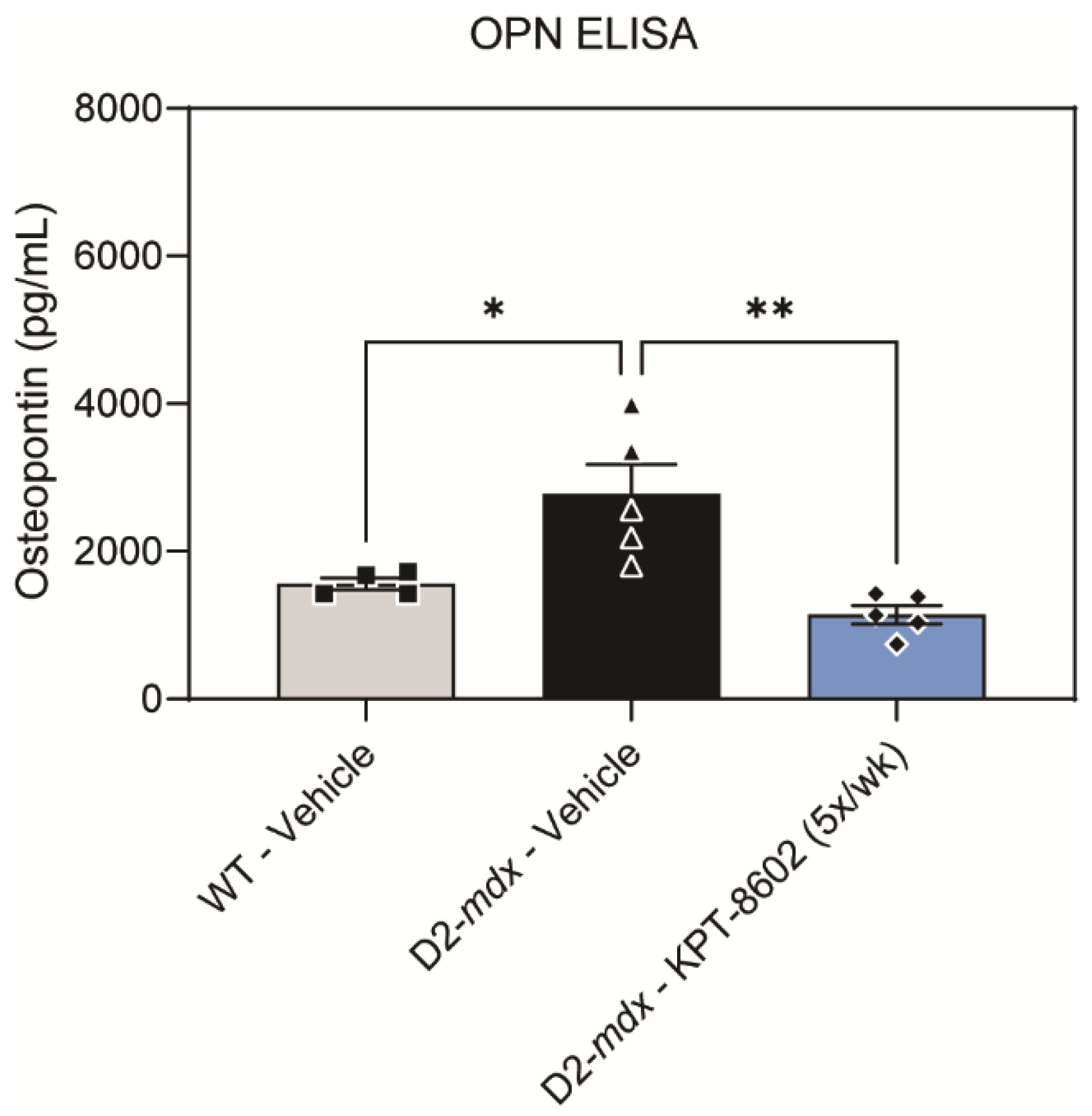
3.4. KPT−8602 Reduced Osteopontin Serum Levels in D2-Mdx Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, I.; Brengman, J.M.; Engel, A.G. Analysis of cytokine expression in muscle in inflammatory myopathies, Duchenne dystrophy, and non-weak controls. J. Neuroimmunol. 1995, 63, 9–16. [Google Scholar] [CrossRef]
- Ishizaki, M.; Suga, T.; Kimura, E.; Shiota, T.; Kawano, R.; Uchida, Y.; Uchino, K.; Yamashita, S.; Maeda, Y.; Uchino, M. Mdx respiratory impairment following fibrosis of the diaphragm. Neuromuscul. Disord. 2008, 18, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv294. [Google Scholar] [CrossRef] [PubMed]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Libr. Cochrane Rev. 2008, 2004, CD003725. [Google Scholar] [CrossRef]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Ricotti, V.; Ridout, D.A.; Scott, E.; Quinlivan, R.; Robb, S.A.; Manzur, A.Y.; Muntoni, F. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 2013, 84, 698–705. [Google Scholar] [CrossRef]
- Anthony, K.; Arechavala-Gomeza, V.; Ricotti, V.; Torelli, S.; Feng, L.; Janghra, N.; Tasca, G.; Guglieri, M.; Barresi, R.; Armaroli, A.; et al. Biochemical characterization of patients with in-frame or out-of-frame dmd deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. 2014, 71, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Rahimov, F.; Kunkel, L.M. Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGFbeta in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Mojumdar, K.; Liang, F.; Lemaire, C.; Li, T.; Richardson, J.; Divangahi, M.; Qureshi, S.; Petrof, B.J. Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 24, 2147–2162. [Google Scholar] [CrossRef] [PubMed]
- Ying Wang, M.W.-H.; Samengo, G.; Tidball, J.G. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 2015, 14, 678–688. [Google Scholar] [CrossRef]
- Kabachinski, G.; Schwartz, T.U. The nuclear pore complex--structure and function at a glance. J. Cell Sci. 2015, 128, 423–429. [Google Scholar] [CrossRef]
- Alexander, T.B.; Lacayo, N.J.; Choi, J.K.; Ribeiro, R.C.; Pui, C.-H.; Rubnitz, J.E. Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Combination with Fludarabine and Cytarabine, in Pediatric Relapsed or Refractory Acute Leukemia. J. Clin. Oncol. 2016, 34, 4094–4101. [Google Scholar] [CrossRef]
- Azizian, N.G.; Li, Y. XPO1-dependent nuclear export as a target for cancer therapy. J. Hematol. Oncol. 2020, 13, 61. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 335–345. [Google Scholar] [CrossRef]
- Kuruvilla, J.; Savona, M.; Baz, R.; Mau-Sorensen, P.M.; Gabrail, N.; Garzon, R.; Stone, R.; Wang, M.; Savoie, L.; Martin, P.; et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017, 129, 3175–3183. [Google Scholar] [CrossRef]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)—A novel class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Razak, A.R.A.; Mau-Soerensen, M.; Gabrail, N.Y.; Gerecitano, J.F.; Shields, A.F.; Unger, T.J.; Saint-Martin, J.R.; Carlson, R.; Landesman, Y.; McCauley, D.; et al. First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4142–4150. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gery, S.; Sun, H.; Shacham, S.; Kauffman, M.; Koeffler, H.P. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2014, 74, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.; Sanglier, J.-J.; Wang, Y. Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 1997, 4, 139–147. [Google Scholar] [CrossRef]
- Callanan, M.; Kudo, N.; Gout, S.; Brocard, M.; Yoshida, M.; Dimitrov, S.; Khochbin, S. Developmentally regulated activity of CRM1/XPO1 during early Xenopus embryogenesis. J. Cell Sci. 2000, 113, 451–459. [Google Scholar] [CrossRef]
- Crochiere, M.; Kashyap, T.; Kalid, O.; Shechter, S.; Klebanov, B.; Senapedis, W.; Saint-Martin, J.-R.; Landesman, Y. Deciphering mechanisms of drug sensitivity and resistance to Selective Inhibitor of Nuclear Export (SINE) compounds. BMC Cancer 2015, 15, 910. [Google Scholar] [CrossRef]
- Crochiere, M.L.; Baloglu, E.; Klebanov, B.; Donovan, S.; Alamo, D.d.; Lee, M.; Kauffman, M.; Shacham, S.; Landesman, Y. A method for quantification of exportin-1 (XPO1) occupancy by Selective Inhibitor of Nuclear Export (SINE) compounds. Oncotarget 2015, 7, 1863–1877. [Google Scholar] [CrossRef]
- Hightower, R.M.; Reid, A.L.; Gibbs, D.E.; Wang, Y.; Widrick, J.J.; Kunkel, L.M.; Kastenschmidt, J.M.; Villalta, S.A.; van Groen, T.; Chang, H.; et al. The SINE Compound KPT-350 Blocks Dystrophic Pathologies in DMD Zebrafish and Mice. Mol. Ther. 2020, 28, 189–201. [Google Scholar] [CrossRef]
- Walker, J.S.; Hing, Z.A.; Harrington, B.; Baumhardt, J.; Ozer, H.G.; Lehman, A.; Giacopelli, B.; Beaver, L.; Williams, K.; Skinner, J.N.; et al. Recurrent XPO1 mutations alter pathogenesis of chronic lymphocytic leukemia. J. Hematol. Oncol. 2021, 14, 17. [Google Scholar] [CrossRef]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S.; et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef]
- Vercruysse, T.; De Bie, J.; Neggers, J.E.; Jacquemyn, M.; Vanstreels, E.; Schmid-Burgk, J.L.; Hornung, V.; Baloglu, E.; Landesman, Y.; Senapedis, W.; et al. The Second-Generation Exportin-1 Inhibitor KPT-8602 Demonstrates Potent Activity against Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 2528–2541. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, D.; Demeyer, S.; Prieto, C.; de Bock, C.E.; De Bie, J.; Gielen, O.; Jacobs, K.; Mentens, N.; Verhoeven, B.M.; Uyttebroeck, A.; et al. The XPO1 Inhibitor KPT-8602 Synergizes with Dexamethasone in Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 5747–5758. [Google Scholar] [CrossRef] [PubMed]
- Hays, J.; Zhang, J.; Berlin, J.D.; O’Hara, M.; Shah, M.A.; Reichmann, W.; Senapedis, W.; Achour, H.; Baloglu, E.; Shacham, S.; et al. Eltanexor (KPT-8602), a second-generation selective inhibitor of nuclear export (SINE) compound, in patients with metastatic colorectal cancer (mCRC). Ann. Oncol. 2018, 29, viii716. [Google Scholar] [CrossRef]
- Smith, L.L.; Beggs, A.H.; Gupta, V.A. Analysis of Skeletal Muscle Defects in Larval Zebrafish by Birefringence and Touch-evoke Escape Response Assays. J. Vis. Exp. 2013, 82, e50925. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Sztal, T.; Currie, P.D. Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys Res. Commun. 2012, 423, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Zaleska, M.M.; Riddell, D.R.; Atchison, K.P.; Robshaw, A.; Zhou, H.; Sukoff Rizzo, S.J. Alternative method of oral administration by peanut butter pellet formulation results in target engagement of BACE1 and attenuation of gavage-induced stress responses in mice. Pharmacol. Biochem. Behav. 2014, 126, 28–35. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Kastenschmidt, J.M.; Avetyan, I.; Villalta, S.A. Characterization of the Inflammatory Response in Dystrophic Muscle Using Flow Cytometry. In Duchenne Muscular Dystrophy: Methods and Protocols; Bernardini, C., Ed.; Springer: New York, NY, USA, 2018; pp. 43–56. [Google Scholar] [CrossRef]
- Coley, W.D.; Bogdanik, L.; Vila, M.C.; Yu, Q.; Van Der Meulen, J.H.; Rayavarapu, S.; Novak, J.S.; Nearing, M.; Quinn, J.L.; Saunders, A.; et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2016, 25, 130–145. [Google Scholar] [CrossRef]
- van Putten, M.; Putker, K.; Overzier, M.; Adamzek, W.A.; Pasteuning-Vuhman, S.; Plomp, J.J.; Aartsma-Rus, A. Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J. 2019, 33, 8110–8124. [Google Scholar] [CrossRef]
- Hing, Z.A.; Fung, H.Y.J.; Ranganathan, P.; Mitchell, S.; El-Gamal, D.; Woyach, J.A.; Williams, K.; Goettl, V.M.; Smith, J.; Yu, X.; et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016, 30, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- Hammers, D.W.; Hart, C.C.; Matheny, M.K.; Wright, L.A.; Armellini, M.; Barton, E.R.; Sweeney, H.L. The D2.mdx mouse as a preclinical model of the skeletal muscle pathology associated with Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 14070. [Google Scholar] [CrossRef]
- Spaulding, H.R.; Quindry, T.; Hammer, K.; Quindry, J.C.; Selsby, J.T. Nutraceutical and pharmaceutical cocktails did not improve muscle function or reduce histological damage in D2-mdx mice. J. Appl. Physiol. 2019, 127, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Coker, C.C.; Jirousek, M.R.; Zimmer, M.; Walter, G.A.; Sweeney, H.L. Disease-modifying effects of orally bioavailable NF-κB inhibitors in dystrophin-deficient muscle. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Yin, X.; Tang, Y.; Li, J.; Dzuricky, A.T.; Pu, C.; Fu, F.; Wang, B. Genetic ablation of P65 subunit of NF-κB in mdx mice to improve muscle physiological function. Muscle Nerve 2017, 56, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kumagai-Cresse, C.; Ermolova, N.; Mokhonova, E.; Marinov, M.; Capote, J.; Becerra, D.; Quattrocelli, M.; Crosbie, R.H.; Welch, E.; et al. Spp1 (osteopontin) promotes TGFβ processing in fibroblasts of dystrophin-deficient muscles through matrix metalloproteinases. Hum. Mol. Genet. 2019, 28, 3431–3442. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Mokhonova, E.I.; Kendall, G.C.; Becerra, D.; Naeini, Y.B.; Cantor, R.M.; Spencer, M.J.; Nelson, S.F.; Miceli, M.C. Repurposing Dantrolene for Long-Term Combination Therapy to Potentiate Antisense-Mediated DMD Exon Skipping in the mdx Mouse. Mol. Ther.—Nucleic Acids 2018, 11, 180–191. [Google Scholar] [CrossRef]
- Li, J.; Fredericks, M.; Cannell, M.; Wang, K.; Sako, D.; Maguire, M.C.; Grenha, R.; Liharska, K.; Krishnan, L.; Bloom, T.; et al. ActRIIB:ALK4-Fc alleviates muscle dysfunction and comorbidities in murine models of neuromuscular disorders. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2008, 18, 482–496. [Google Scholar] [CrossRef]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.L.; Carathers, M.; Li, Z.-W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenberg, A.S.; Bluestone, J.A. The immune system in Duchenne muscular dystrophy: Friend or foe. Rare Dis. 2015, 3, e1010966. [Google Scholar] [CrossRef] [Green Version]
- Dahiya, S.; Givvimani, S.; Bhatnagar, S.; Qipshidze, N.; Tyagi, S.C.; Kumar, A. Osteopontin-Stimulated Expression of Matrix Metalloproteinase-9 Causes Cardiomyopathy in the mdx Model of Duchenne Muscular Dystrophy. J. Immunol. 2011, 187, 2723–2731. [Google Scholar] [CrossRef]
- Vetrone, S.A.; Montecino-Rodriguez, E.; Kudryashova, E.; Kramerova, I.; Hoffman, E.P.; Liu, S.D.; Miceli, M.C.; Spencer, M.J. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-β. J. Clin. Investig. 2009, 119, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, M.; Kimura, E.; Nagata, T.; Okada, T.; Aoki, Y.; Tachimori, H.; Yonemoto, N.; Imamura, M.; Takeda, S.i. Serum Osteopontin as a Novel Biomarker for Muscle Regeneration in Duchenne Muscular Dystrophy. Am. J. Pathol. 2016, 186, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, E.; Hoffman, E.P.; Piva, L.; Gavassini, B.F.; Cagnin, S.; Ermani, M.; Bello, L.; Soraru, G.; Pacchioni, B.; Bonifati, M.D.; et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology 2011, 76, 219–226. [Google Scholar] [CrossRef] [PubMed]
- van den Bergen, J.C.; Hiller, M.; Böhringer, S.; Vijfhuizen, L.; Ginjaar, H.B.; Chaouch, A.; Bushby, K.; Straub, V.; Scoto, M.; Cirak, S.; et al. Validation of genetic modifiers for Duchenne muscular dystrophy: A multicentre study assessing SPP1 and LTBP4 variants. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Capote, J.; Kramerova, I.; Martinez, L.; Vetrone, S.; Barton, E.R.; Sweeney, H.L.; Miceli, M.C.; Spencer, M.J. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol. 2016. [Google Scholar] [CrossRef]
- Conforti, F.; Zhang, X.; Rao, G.; De Pas, T.; Yonemori, Y.; Rodriguez, J.A.; McCutcheon, J.N.; Rahhal, R.; Alberobello, A.T.; Wang, Y.; et al. Therapeutic Effects of XPO1 Inhibition in Thymic Epithelial Tumors. Cancer Res. 2017, 77, 5614–5627. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
English, K.G.; Reid, A.L.; Samani, A.; Coulis, G.J.F.; Villalta, S.A.; Walker, C.J.; Tamir, S.; Alexander, M.S. Next-Generation SINE Compound KPT−8602 Ameliorates Dystrophic Pathology in Zebrafish and Mouse Models of DMD. Biomedicines 2022, 10, 2400. https://doi.org/10.3390/biomedicines10102400
English KG, Reid AL, Samani A, Coulis GJF, Villalta SA, Walker CJ, Tamir S, Alexander MS. Next-Generation SINE Compound KPT−8602 Ameliorates Dystrophic Pathology in Zebrafish and Mouse Models of DMD. Biomedicines. 2022; 10(10):2400. https://doi.org/10.3390/biomedicines10102400
Chicago/Turabian StyleEnglish, Katherine G., Andrea L. Reid, Adrienne Samani, Gerald J. F. Coulis, S. Armando Villalta, Christopher J. Walker, Sharon Tamir, and Matthew S. Alexander. 2022. "Next-Generation SINE Compound KPT−8602 Ameliorates Dystrophic Pathology in Zebrafish and Mouse Models of DMD" Biomedicines 10, no. 10: 2400. https://doi.org/10.3390/biomedicines10102400
APA StyleEnglish, K. G., Reid, A. L., Samani, A., Coulis, G. J. F., Villalta, S. A., Walker, C. J., Tamir, S., & Alexander, M. S. (2022). Next-Generation SINE Compound KPT−8602 Ameliorates Dystrophic Pathology in Zebrafish and Mouse Models of DMD. Biomedicines, 10(10), 2400. https://doi.org/10.3390/biomedicines10102400