2.1. General Information
Unless noted otherwise, reactants and reagents were purchased from commercial sources and used without further purification. Dry CH
2Cl
2, Et
2O, THF and MeOH were obtained from a dispensing system by passing commercial material through a cartridge containing activated alumina (PURESOLV, Innovative Technology, Oldham, UK), stored under dry nitrogen, and then used as such without further drying unless specified.
DMSO was dried by treating commercial material with CaH
2 mesh at 150 °C under argon, followed by distillation under reduced pressure [
14]. Deoxygenated and dry THF was obtained by refluxing and distilling pre-dried material (as described above) from sodium and benzophenone under argon.
Molecular sieves were activated by heating them to 200 °C for approximately 6 h in a high vacuum and were then stored under argon [
15].
Melting ranges were determined using a Kofler-type Leica Galen III micro hot stage microscope or an SRS OptiMelt Automated Melting Point System and are uncorrected. Temperatures are reported in intervals of 0.5 °C.
Aluminum-backed Merck silica gel 60 with the fluorescence indicator F254 was used for thin layer chromatography (TLC). Spots were visualized under UV light (254 nm) and by staining with cerium ammonium molybdate (CAM) solution (20 g of ammonium pentamolybdate, 0.8 g of cerium(IV) ammonium sulfate, 400 mL of 10 v/v % sulfuric acid) as a general purpose reagent. Alcohols were also visualized with p-anisaldehyde solution (3.5 g p-anisaldehyde, 1.5 mL acetic acid, 5 mL sulfuric acid, 120 mL ethanol), and compounds pertaining double bonds were visualized with potassium permanganate solution (1.5 g potassium permanganate, 10 g potassium carbonate, 1 mL 10 w/w % NaOH, 200 mL water).
Specific rotation was measured using an Anton Parr MCP500 polarimeter (AntonPaar GmbH, Graz, Austria) and HPLC-grade solvents under conditions as specified individually. Values are reported in the form + or—specific rotation (concentration in terms of g/100 mL, solvent).
Analytical chromatography–spectroscopy gas chromatography–mass spectroscopy (GC-MS) was used to analyze samples of reaction products with sufficient volatility. The following instruments and columns were used: Thermo Scientific (Fisher Scientific GmbH, Schwerte, Germany) Finnigan Focus GC/Quadrupole DSQ II device using a helium flow of 2.0 mL/min, analyzing an m/z range from 50 to 650; BGB 5 (0.25 µm film; 30 m × 0.25 mm ID). Temperature gradients are as follows: Method A: 100 °C (2 min), to 280 °C in 10 min, 11 min hold-time at 280 °C (23 min); Method B: 80 °C (2 min), to 280 °C in 10 min (20 °C/min), 12 min hold-time at 280 °C (24 min); Method C: 100 °C (2 min), to 280 °C in 4.5 min (40 °C/min), 16.5 min hold-time at 280 °C (23 min); Method D: 100 °C (2 min), to 280 °C in 4.5 min (40 °C/min), 31.5 min hold-time at 280 °C (38 min); Method E: 100 °C (2 min), to 280 °C in 4.5 min (40 °C/min), 41.5 min hold-time at 280 °C (48 min). Data are reported in the form retention time; m/z1 (relative intensity in %), m/z2 (relative intensity in %), Only signals with m/z ≥ 90 and relative intensity ≥ 15% are given, except for the signal at 100% relative intensity, which is always given. Additionally, the molecular ion signal M+ is given regardless of its intensity or m/z; in cases where M+ was not visible due to excessive fragmentation, a characteristic fragment signal is identified instead.
High-pressure liquid chromatography (HPLC) was used to determine the enantiomeric excess of reaction products, using a Dionex UltiMate 3000 device (RS Diode Array Detector Fisher Scientific GmbH, Schwerte, Germany). Chiral separation columns and analysis conditions are specified individually. In all cases, retention times include appropriate guard cartridges containing the same stationary phase as the separation column.
Liquid chromatography–high-resolution mass spectroscopy (LC-HRMS) was used to confirm the exact molecular mass of reaction products by their quasi-molecular ions (M + H+ or M + Na+). The following two instruments were used: (1) Shimadzu Prominence HPLC device (DGU-20 A3 degassing unit, 2 × LC20AD binary gradient pump, SIL-20 A auto injector, CTO-20AC column oven, CBM-20A control module, and SPD-M20A diode array detector). Samples were eluted through a Phenomenex Kinetex precolumn (5 μm core shell ODS(3) phase; 4 mm × 2 mm ID) at 40 °C under conditions comprising gradients of H2O/MeOH containing formic acid (0.1 v/v %), and then detected using a Shimadzu IT-TOF-MS by electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI), as indicated individually. Analyses were performed by E. Rosenberg (CTA, VUT) and L. Czollner (IAS, VUT); (2) Agilent 1100/1200 HPLC device (degassing unit, 1200SL binary gradient pump, column thermostat, and CTC Analytics HTC PAL autosampler). Samples were eluted through a silica-based Phenomenex C-18 Security guard cartridge (1.7 µm PD; 2.1 mm ID) at 40 °C under isocratic conditions comprising H2O containing formic acid (0.1 v/v %)/MeOH containing formic acid (0.1 v/v %) in a ratio of 30:70 at a flow rate of 0.5 mL/min, and then detected using an Agilent 6230 LC-TOF-MS equipped with an Agilent Dual AJS ESI source by electrospray ionization (ESI). Analyses were performed by L. Czollner (IAS, VUT). Flash column chromatography was carried out on Merck silica gel 60 (40–63 μm), and separations were performed using a Büchi Sepacore system (dual Pump Module C-605, Pump Manager C-615, Fraction Collector C-660, and UV Monitor C-630 or UV Photometer C635).
Preparative high-performance liquid chromatography (preparative HPLC) was carried out on a Phenomenex Luna reverse-phase column (10 µm C18(2) phase, 100 A; 250 mm × 21.20 mm ID), and separations were performed using a Shimadzu LC-8A device (SIL-10AP autosampler, SPD-20 detector, and FRC-10A fraction collector (Shimadzu Österreich, Korneuburg, Austria). Reaction temperatures were measured externally (electronic thermometer connected to a heater–stirrer or low-temperature thermometer in the case of cryogenic reactions), unless otherwise noted.
Nuclear magnetic resonance spectroscopy (NMR) spectra were recorded from CDCl
3 or DMSO-d6 solutions on a Bruker AC 200 (200 MHz proton resonance frequency) or a Bruker Advanced UltraShield (400 MHz) spectrometer (as indicated individually) from Bruker Daltonik GmbH, Bremen, Germany, and chemical shifts are reported in ascending order in ppm relative to the nominal residual solvent signals, i.e., 1H: δ = 2.50 ppm (DMSO-d6); 13C: δ = 77.16 ppm (CDCl
3), δ = 39.52 ppm (DMSO-d6) [
5,
6]. For all 1H spectra in CDCl
3, however, shifts are reported relative to tetramethyl silane (TMS) as an internal standard (δ = 0 ppm) due to the interference of aromatic signals of many samples with the residual solvent signal of CDCl
3. For the 13C spectra, the J-modulated attached proton test (APT) or distortionless enhancement by polarization transfer (DEPT-135) pulse sequences were used to aid in the assignment.
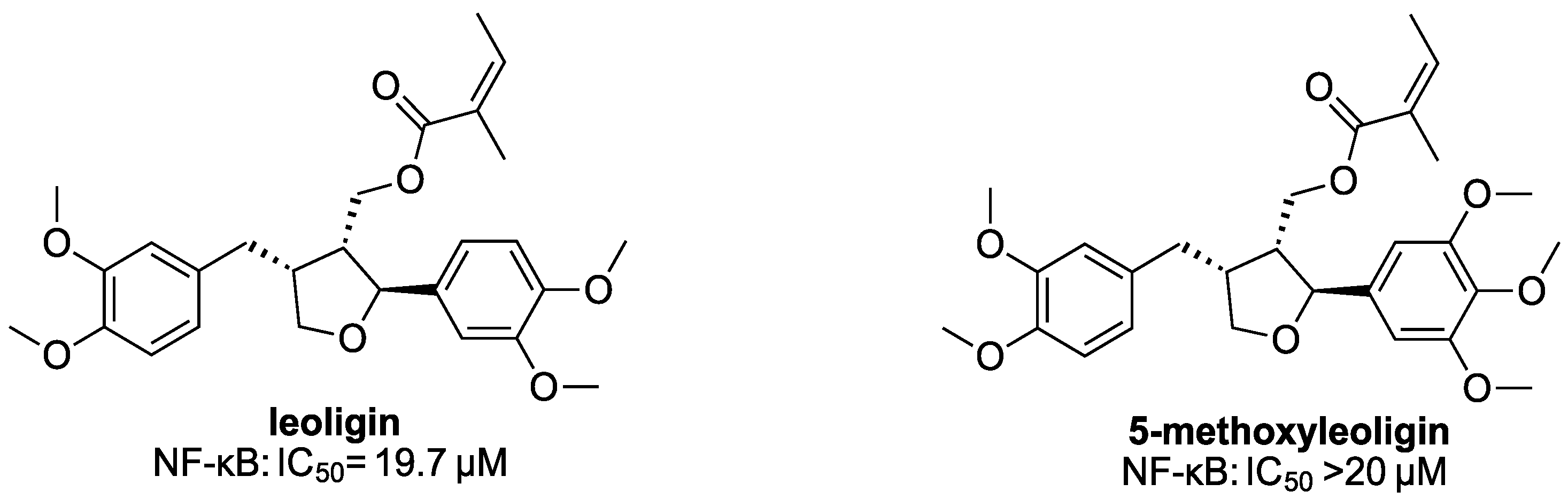
NF-κB transactivation activity was determined by a luciferase reporter gene assay in HEK293 cells stably transfected with the NF-κB-driven luciferase reporter gene NF-κB-luc (293/NF-κB-luc cells, Panomics, Fremont, CA, USA, RC0014) [
16]. Cells were stained with 2 µM cell tracker green (CTG, Thermo Scientific). After one hour, 4 × 10⁴ cells per well were seeded in a 96-well plate in serum-free DMEM (4.5 g/L glucose) obtained from Lonza and supplemented with 2 mM glutamine, 100 U/mL benzylpenicillin and 100 µg/mL streptomycin. After incubation at 37 °C, 5% CO
2 overnight, cells were pre-treated on the next day with test samples for 1 h. Thereafter, cells were stimulated with 2 ng/mL human recombinant TNF-α (Sigma-Aldrich Handels Gmbh, Vienna, Austria) for 4 h to activate the NF-κB signaling pathway. Then, the medium was removed, and cells were lysed with luciferase reporter lysis buffer (E3971, Promega, Madison, WI, USA). Leoligin and its derivatives were tested at a concentration of 20 µM (screening) in at least three independent experiments. The sesquiterpene lactone parthenolide, an effective inhibitor of the NF-κB pathway [
12], was used as a positive control at a concentration of 5 µM and 0.1% DMSO served as vehicle control. The luminescence of the firefly luciferase product and the CTG-derived fluorescence were quantified on a Tecan GeniosPro plate reader (Tecan, Grödig, Austria). The ratio of luminescence units to fluorescence units was calculated to account for differences in cell number. Results were expressed as fold changed relative to the vehicle control with TNFα, which was set to 1 [
16]. CTG-fluorescence values used to estimate cell viability were also normalized to the vehicle control with TNFα. Compared to the vehicle control, treatments with fluorescence values below 0.75 were considered as toxic.
IC50 values were determined by the luciferase reporter assay, as described above. Dose–response curves were established by measuring compounds at concentrations of 20 µM, 10 µM, 5 µM and 1 µM, each concentration in three independent experiments. For statistical analyses, the IC50 values were calculated using nonlinear regression, with sigmoidal dose responses (GraphPad Software 4.03., Inc., San Diego, CA, USA).
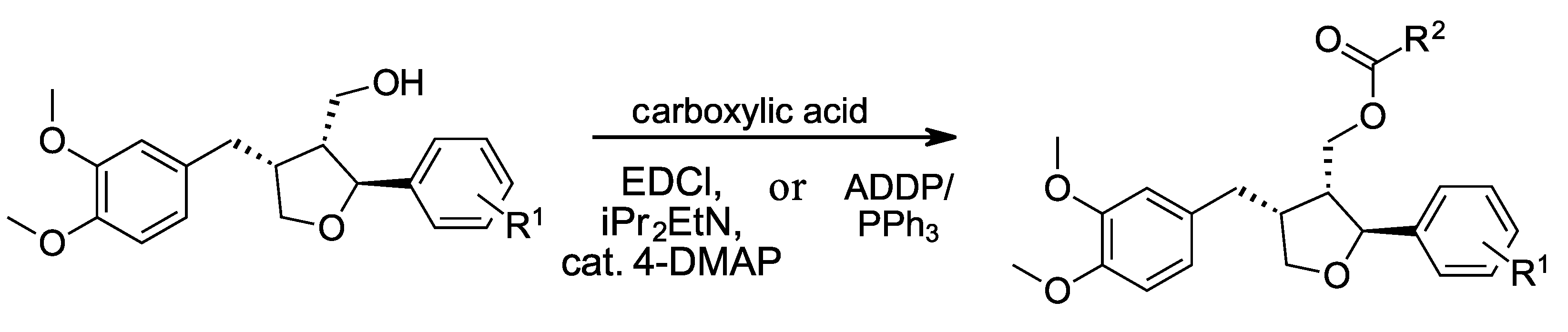
2.2. Syntheses of Target Compounds via Steglich Esterification
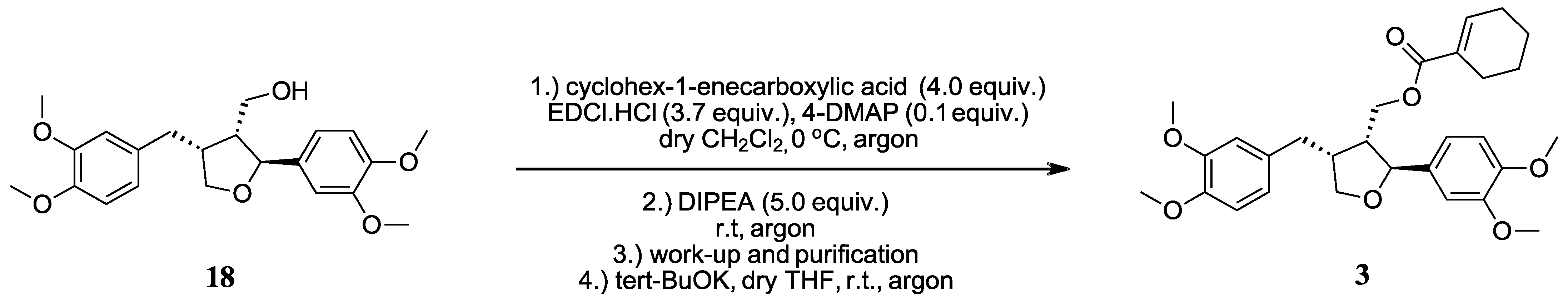
2.2.1. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(3,4-dimethoxyphenyl)tetrahydrofuran-3-yl)methyl cyclohex-1-enecarboxylate (3)
A reaction vessel was charged with a stirring bar, cyclohex-1-enecarboxylic acid (45.4 mg, 0.360 mmol, 4.0 equiv.) and 4-DMAP (1.1 mg, 9.0 μmol, 0.1 equiv.), and then evacuated and back-filled with argon using standard Schlenk technique. Dry CH
2Cl
2 (1.0 mL) was then added via syringe and the solution was cooled to 0 °C in an ice bath. The vessel was briefly opened, EDCI.HCl (63.8 mg, 0.333 mmol, 3.7 equiv.) was added in one go, and the mixture was stirred for 3 h at 0 °C. Meanwhile, a second vessel was charged with a stirring bar and starting material
18 (35.0 mg, 0.090 mmol, 1.00 equiv.), evacuated and back-filled with argon (3×), and DIPEA (78 μL, 0.45 mmol, 5.0 equiv.) was added via syringe. After 3 h, the solution containing the activated carboxylic acid was transferred to the second vial via syringe and stirred for 24 h at room temperature. The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 10:90 to 22:78 in 9 min, then 22:78 isocratically for 6 min, then to 62:38 in 30 min) to afford the title compound
3 with an overall yield of 60% as a colorless oil (
Scheme 1). [α]
D20: +20.5 (c 2.64, MeOH) LC-HRMS (ESI): calculated for M + Na
+: 519.2353, found: 519.2378, Δ: 4.81 ppm. (log
P)
calc: 5.95 ± 0.45. GC-MS (EI, 70 eV, Method E): 48.95 min; 496.3 (M
+, 1), 370.2 (15), 219.1 (32), 207.0 (24), 206.1 (16), 189.1 (17), 177.1 (17), 166.1 (15), 165.1 (89), 152.1 (16), 151.1 (100), 109.1 (39), 107.1 (19).
1H NMR (200 MHz, CDCl
3): δ 1.51–1.71 (m, 4H), 2.10–2.27 (m, 4H), 2.50–2.95 (m, 4H), 3.76 (dd,
2J = 8.5 Hz,
3J = 6.2 Hz, 1H), 3.86 (s, 3H), 3.87 (s, 6H), 3.88 (s, 3H), 4.08 (dd,
2J = 8.5 Hz,
3J = 6.2 Hz, 1H), 4.26 (dd,
2J = 11.3 Hz,
3J = 7.1 Hz, 1), 4.42 (dd,
2J = 11.3 Hz,
3J = 6.5 Hz, 1H), 4.83 (d,
3J = 6.3 Hz, 1H), 6.67–6.92 (m, 7H).
13C NMR (50 MHz, CDCl
3): δ 21.5 (d), 22.1 (d), 24.2 (d), 25.9 (d), 33.4 (t), 42.8 (d), 49.2 (d), 56.00 (q), 56.02 (q), 56.03 (q), 56.04 (q), 62.6 (t), 72.9 (t), 83.3 (d), 109.1 (d), 111.1 (d), 111.4 (d), 112.0 (d), 118.3 (d), 120.6 (d), 130.1 (s), 132.8 (s), 135.1 (s), 140.5 (d), 147.6 (s), 148.5 (s), 149.1 (s), 149.1 (s), 167.5 (s).
2.2.2. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(3,4-dimethoxyphenyl)tetrahydrofuran-3-yl)methyl cyclobutanecarboxylate (7)
Analogous to 3, using starting material 18 (21.5 mg, 0.055 mmol, 1.00 equiv.), cyclobutanecarboxylic acid (12.7 mg, 0.127 mmol, 2.3 equiv.), EDCI.HCl (21.2 mg, 0.111 mmol, 2.0 equiv.), 4-DMAP (0.7 mg, 5.5 µmol, 0.1 equiv.) and DIPEA (24 µL, 0.14 mmol, 2.5 equiv.), and stirring the reaction for 24 h. The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 10:90 to 15:85 in 10 min, then to 25:75 in 5 min, then to 50:50 in 7 min) to afford the title compound 7 with an overall yield of 83% as a colorless oil. [α]D23: +22.8 (c 1.27, MeOH). LC-HRMS (APCI): calculated for M + Na+: 493.2197, found: 493.2214, Δ: 3.45 ppm. (log P)calc: 4.62 ± 0.44. GC-MS (EI, 70 eV, Method D): 22.60 min; 470.1 (M+, 4), 207.0 (52), 177.1 (16), 166.1 (17), 165.1 (50), 151.0 (100), 107.1 (15). 1H NMR (200 MHz, CDCl3): δ 1.79–2.06 (m, 2H), 2.07–2.37 (m, 4H), 2.47–2.93 (m, 4H), 3.11 (quint, 3J = 8.6 Hz, 1H), 3.75 (dd, 2J = 8.5 Hz, 3J = 6.3 Hz, 1H), 3.86 (s, 3H), 3.87 (s, 6H), 3.89 (s, 3H), 4.07 (dd, 2J = 8.5 Hz, 3J = 6.3 Hz, 1H), 4.19 (dd, 2J = 11.2 Hz, 3J = 7.1 Hz, 1H), 4.38 (dd, 2J = 11.2 Hz, 3J = 6.9 Hz, 1H), 4.80 (d, 3J = 6.4 Hz, 1H), 6.66–6.90 (m, 6H). 13C NMR (50 MHz, CDCl3): δ 18.6 (t), 25.4 (t), 25.4 (t), 33.3 (t), 38.2 (d), 42.7 (d), 49.2 (d), 56.0 (q), 62.6 (t), 72.9 (t), 83.1 (d), 109.0 (d), 111.1 (d), 111.5 (d), 112.0 (d), 118.2 (d), 120.6 (d), 132.7 (s), 135.1 (s), 147.6 (s), 148.6 (s), 149.1 (s), 149.2 (s), 175.4 (s).
2.2.3. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(3,4-dimethoxyphenyl)tetrahydrofuran-3-yl)methylbutyrate (8)
Analogous to 3, using starting material 18 (35.0 mg, 0.090 mmol, 1.00 equiv.), butyric acid (18.2 mg, 0.207 mmol, 2.3 equiv.), EDCI.HCl (34.5 mg, 0.180 mmol, 2.0 equiv.), 4-DMAP (1.1 mg, 9.0 µmol, 0.1 equiv.) and DIPEA (39 µL, 0.23 mmol, 2.5 equiv.). The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 10:90 to 22:78 in 9 min, then 22:78 isocratically for 6 min, then to 62:38 in 30 min) to afford the title compound 8 with an overall yield of 85% as a colorless oil. [α]D20: +20.3 (c 1.77, MeOH). LC-HRMS (ESI): calculated for M + Na+: 481.2197, found: 481.2207, Δ: 2.08 ppm. (log P)calc:4.75 ± 0.44. GC-MS (EI, 70 eV, Method D): 20.50 min; 458.2 (M+, 6), 219.1 (21), 165.1 (52), 152.1 (15), 151.1 (100). 1H NMR (200 MHz, CDCl3): δ 0.94 (t, 3J = 7.4 Hz, 3H), 1.64 (sext, 3J = 7.4 Hz, 2H), 2.26 (t, 3J = 7.4 Hz, 2H), 2.47–2.93 (m, 4H), 3.75 (dd, 2J = 8.6 Hz, 3J = 6.2 Hz, 1H), 3.86 (s, 3H), 3.87 (s, 6H), 3.89 (s, 3H), 4.07 (dd, 2J = 8.6 Hz, 3J = 6.3 Hz, 1H), 4.19 (dd, 2J = 11.2 Hz, 3J = 7.1 Hz, 1H), 4.38 (dd, 2J = 11.2 Hz, 3J = 6.9 Hz, 1H), 4.79 (d, 3J = 6.4 Hz, 1H), 6.64–6.92 (m, 6H). 13C NMR (50 MHz, CDCl3): δ 13.8 (q), 18.5 (t), 33.3 (t), 36.3 (t), 42.6 (d), 49.1 (d), 56.0 (q), 62.5 (t), 72.8 (t), 83.0 (d), 109.0 (d), 111.1 (d), 111.4 (d), 112.0 (d), 118.2 (d), 120.5 (d), 132.7 (s), 135.0 (s), 147.6 (s), 148.6 (s), 149.0 (s), 149.1 (s), 173.6 (s).
2.2.4. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(3,4-dimethoxyphenyl)tetrahydrofuran-3-yl)methyl 3,3-dimethylbutanoate (10)
Analogous to 3, using starting material 18 (30.0 mg, 0.077 mmol, 1.00 equiv.) and 3,3-dimethylbutanoic acid (20.6 mg, 0.178 mmol, 2.3 equiv.), EDCI.HCl (29.6 mg, 0.154 mmol, 2.0 equiv.), 4-DMAP (0.9 mg, 7.7 µmol, 0.1 equiv.) and DIPEA (34 µL, 0.19 mmol, 2.5 equiv.). The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 10:90 to 30:70 in 30 min) to afford the title compound 10 with an overall yield of 51% as a colorless oil. [α]D23: +24.7 (c 1.01, MeOH). LC-HRMS (APCI): calculated for M+Na+: 509.2510, found: 509.2512, Δ: 0.39 ppm. (log P)calc: 5.45 ± 0.45. GC-MS (EI, 70 eV, Method D): 18.75 min; 486.1 (M+, 4), 219.0 (17), 166.0 (17), 165.0 (45), 152.1 (15), 151.0 (100). 1H NMR (200 MHz, CDCl3): δ 1.03 (s, 9H), 2.20 (s, 2H), 2.47–2.63 (m, 2H), 2.64–2.82 (m, 1H), 2.88 (dd, 2J = 12.5 Hz, 3J = 4.2 Hz, 1H), 3.76 (dd, 2J = 8.6 Hz, 3J = 6.1 Hz, 1H), 3.86 (s, 3H), 3.87 (s, 3H), 3.87 (s, 3H), 3.89 (s, 3H), 4.06 (dd, 2J = 8.6 Hz, 3J = 6.2 Hz, 1H), 4.16 (dd, 2J = 11.3 Hz, 3J = 6.9 Hz, 1H), 4.36 (dd, 2J = 11.3 Hz, 3J = 7.2 Hz, 1H), 4.80 (d, 3J = 6.4 Hz, 1H), 6.66–6.90 (m, 6H). 13C NMR (50 MHz, CDCl3): δ 29.8 (q), 30.9 (s), 33.2 (t), 42.6 (d), 48.1 (t), 49.3 (d), 56.0 (q), 62.3 (t), 72.8 (t), 82.9 (d), 109.0 (d), 111.2 (d), 111.5 (d), 112.0 (d), 118.2 (d), 120.6 (d), 132.7 (s), 135.1 (s), 147.7 (s), 148.6 (s), 149.1 (s), 149.2 (s), 172.4 (s).
2.2.5. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(3,4-dimethoxyphenyl)tetrahydrofuran-3-yl)methyl2-ethylbutanoate (11)
Analogous to 3, using starting material 18 (20.3 mg, 0.052 mmol, 1.00 equiv.) and 2-ethylbutanoic acid (14.0 mg, 0.120 mmol, 2.3 equiv.), EDCI.HCl (20.0 mg, 0.105 mmol, 2.0 equiv.), 4-DMAP (0.6 mg, 5.2 µmol, 0.1 equiv.) and DIPEA (23 µL, 0.13 mmol, 2.5 equiv.). The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 10:90 to 30:70 in 30 min) to afford the title compound 11 with an overall yield of 87% as a colorless oil. [α]D23: +23.3 (c 0.88, MeOH).LC-HRMS (APCI): calculated for M + Na+: 509.2510, found: 509.2533, Δ: 4.52 ppm. (log P)calc: 5.63 ± 0.45. GC-MS (EI, 70 eV, Method E): 23.47 min; 486.2 (M+, 7), 219.1 (32), 189.1 (15), 165.1 (56), 151.0 (100). 1H NMR (200 MHz, CDCl3): δ 0.90 (t, 3J = 7.4 Hz, 6H), 1.42–1.75 (m, 4H), 2.15–2.30 (m, 1H), 2.47–2.82 (m, 3H), 2.88 (dd, 2J = 12.6 Hz, 3J = 4.0 Hz, 1H), 3.76 (dd, 2J = 8.6 Hz, 3J = 6.1 Hz, 1H), 3.87 (s, 6H), 3.87 (s, 3H), 3.89 (s, 3H), 4.06 (dd, 2J = 8.6 Hz, 3J = 6.2 Hz, 1H), 4.18 (dd, 2J = 11.3 Hz, 3J = 6.8 Hz, 1H), 4.41 (dd, 2J = 11.3 Hz, 3J = 7.1 Hz, 1H), 4.81 (d, 3J = 6.4 Hz, 1H), 6.64–6.91 (m, 6H).
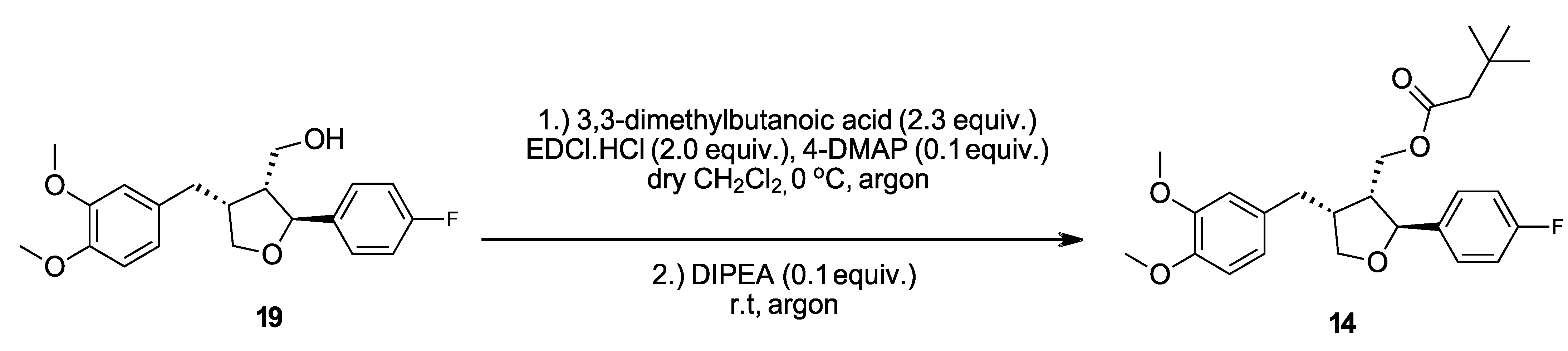
2.2.6. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(4-fluorophenyl)tetrahydrofuran-3-yl)methyl 3,3-dimethylbutanoate (14)
Analogous to
3, using starting material
19 (22.0 mg, 0.064 mmol, 1.00 equiv.), 3,3-dimethylbutanoic acid (16.9 mg, 0.146 mmol, 2.3 equiv.), EDCI.HCl (24.3 mg, 0.127 mmol, 2.0 equiv.), 4-DMAP (0.8 mg, 6.4 µmol, 0.1 equiv.) and DIPEA (28 µL, 0.16 mmol, 2.5 equiv.), and stirring for 13.5 in place of 24 h. The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 9:91 to 25:75 in 60 min) to afford the title compound
14 with an overall yield of 92% as a colorless oil (
Scheme 2). [α]
D23: +15.7 (c 2.07, MeOH). LC-HRMS (APCI): calculated for M + H
+: 445.2385, found: 445.2398, Δ: 2.92 ppm. (log
P)
calc: 5.76 ± 0.50. GC-MS (EI, 70 eV, Method D): 11.19 min; 444.1 (M
+, 7), 194.0 (15), 190.1 (17), 189.1 (19), 177.0 (39), 164.1 (19), 152.0 (28), 151.0 (100), 123.0 (42), 109.0 (24), 107.0 (16).
1H NMR (200 MHz, CDCl
3): δ 1.03 (s, 9H), 2.19 (s, 2H), 2.43–2.62 (m, 2H), 2.62–2.82 (m, 1H), 2.86 (dd,
2J = 12.5 Hz,
3J = 4.2 Hz, 1H), 3.77 (dd,
2J = 8.6 Hz,
3J = 6.3 Hz, 1H), 3.86 (s, 3H), 3.86 (s, 3H), 4.06 (dd,
2J = 8.6 Hz,
3J = 6.2 Hz, 1H), 4.16 (dd,
2J = 11.1 Hz,
3J = 7.2 Hz, 1H), 4.37 (dd,
2J = 11.3 Hz,
3J = 6.9 Hz, 1H), 4.85 (d,
3J = 6.1 Hz, 1H), 6.64–6.84 (m, 3H), 6.96–7.09 (m, 2H), 7.23–7.34 (m, 2H).
13C NMR (50 MHz, CDCl
3): δ 29.8 (q), 30.9 (s), 33.1 (t), 42.6 (d), 48.1 (t), 49.5 (d), 56.00 (q), 56.04 (q), 62.2 (t), 72.9 (t), 82.7 (d), 111.5 (d), 112.0 (d), 115.4 (dd), 120.6 (d), 127.5 (dd), 132.6 (s), 138.5 (d), 147.7 (s), 149.1 (s), 162.4 (d), 172.4 (s).
2.2.7. ((2S,3R,4R)-4-(3,4-Dimethoxybenzyl)-2-(4-fluorophenyl)tetrahydrofuran-3-yl)methyl cyclohexanecarboxylate (17)
Analogous to 3, using starting material 19 (31.2 mg, 0.090 mmol, 1.00 equiv.), cyclohexanecarboxylic acid (26.5 mg, 0.207 mmol, 2.3 equiv.), EDCI.HCl (34.5 mg, 0.180 mmol, 2.0 equiv.), 4-DMAP (1.1 mg, 9.0 µmol, 0.1 equiv.) and DIPEA (39 µL, 0.23 mmol, 2.5 equiv.). The reaction solution was used directly for flash column chromatography (9 g silica, flow rate 20 mL/min, EtOAc/LP, 0:100 to 15:85 in 16 min, then to 22:78 in 4 min) to afford the title compound 17 with an overall yield of 91% as a colorless oil. [α]D20: +15.1 (c 2.10, MeOH). LC-HRMS (ESI): calculated for M+Na+: 479.2204, found: 479.2185, Δ: −3.96 ppm. (log P)calc: 6.06 ± 0.49. GC-MS (EI, 70 eV, Method C): 19.53 min; 456.3 (M+, 4), 194.1 (24), 190.1 (24), 189.1 (23), 177.1 (44), 164.1 (26), 163.1 (16), 152.1 (30), 151.1 (100), 123.0 (47), 109.1 (33), 107.1 (18). 1H NMR (200 MHz, CDCl3): δ 1.11–1.95 (m, 10H), 2.17–2.34 (m, 1H), 2.42–2.62 (m, 2H), 2.62–2.79 (m, 1H), 2.85 (dd, 2J = 12.4 Hz, 3J = 4.3 Hz, 1H), 3.77 (dd, 2J = 8.6 Hz, 3J = 6.3 Hz, 1H), 3.86 (s, 3H), 3.87 (s, 3H), 4.07 (dd, 2J = 8.6 Hz, 3J = 6.3 Hz, 1H), 4.17 (dd, 2J = 11.3 Hz, 3J = 7.3 Hz, 1H), 4.38 (dd, 2J = 11.3 Hz, 3J = 6.7 Hz, 1H), 4.84 (d, 3J = 6.2 Hz, 1H), 6.65–6.75 (m, 2H), 6.81 (d, 3J = 7.9 Hz, 1H), 7.02 (dd, 3J = 8.7 Hz, 3JH-F = 8.7 Hz, 2H), 7.29 (dd, 3J = 8.6 Hz, 4JH-F = 5.4 Hz, 2H). 13C NMR (50 MHz, CDCl3): δ 25.5 (t), 25.8 (t), 29.1 (t), 29.1 (t), 33.2 (t), 42.7 (d), 43.3 (d), 49.5 (d), 55.97 (q), 56.00 (q), 62.3 (t), 72.9 (t), 82.7 (d), 111.4 (d), 111.9 (d), 115.4 (dd), 120.5 (d), 127.5 (dd), 132.6 (s), 138.5 (d), 147.7 (s), 149.1 (s), 162.3 (d), 176.0 (s).



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















