
Micro-RNA 92a as a Therapeutic Target for Cardiac Microvascular Dysfunction in Diabetes

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. EC Culture
2.2. Transfection
2.3. Tube Formation
2.4. Wound Healing
2.5. Flow Chamber Assay
2.6. Proliferation Assay
2.7. Western Blot
2.8. Quantitative PCR
2.9. ImageJ Analysis
2.10. Immunofluorescence
2.11. Statistical Analysis
3. Results
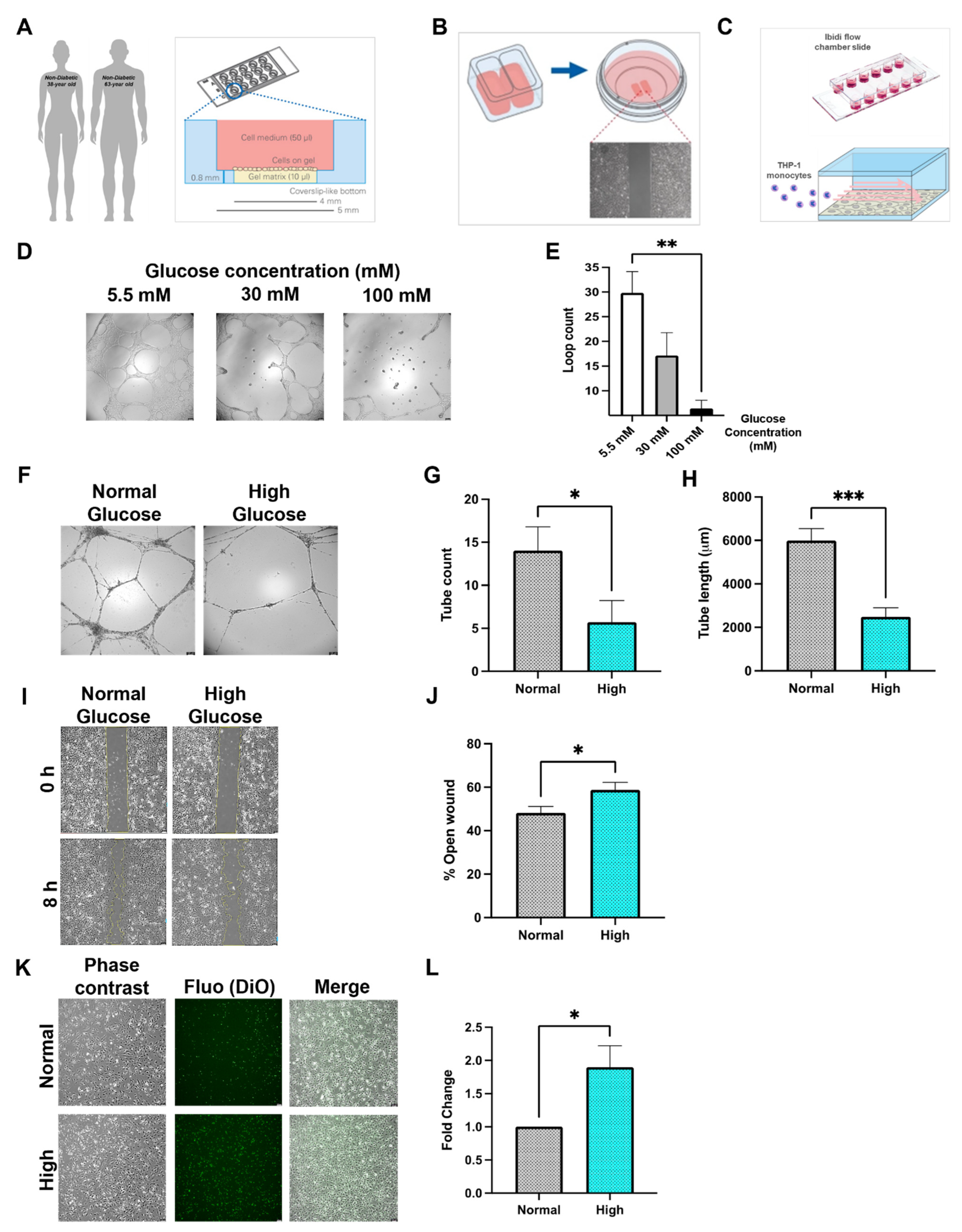
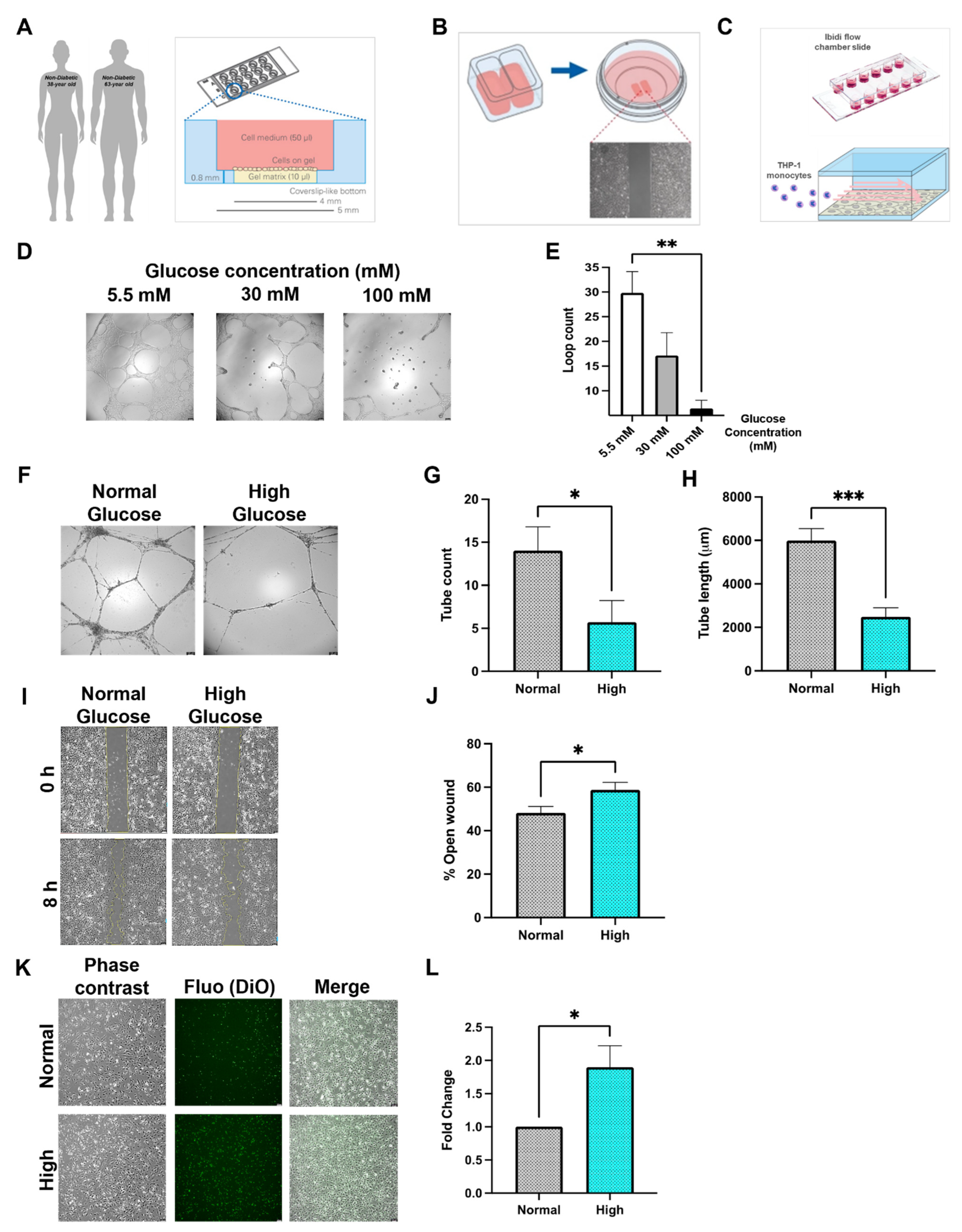
3.1. Diabetes In Vitro and Its Relevance for Microcirculatory EC Function
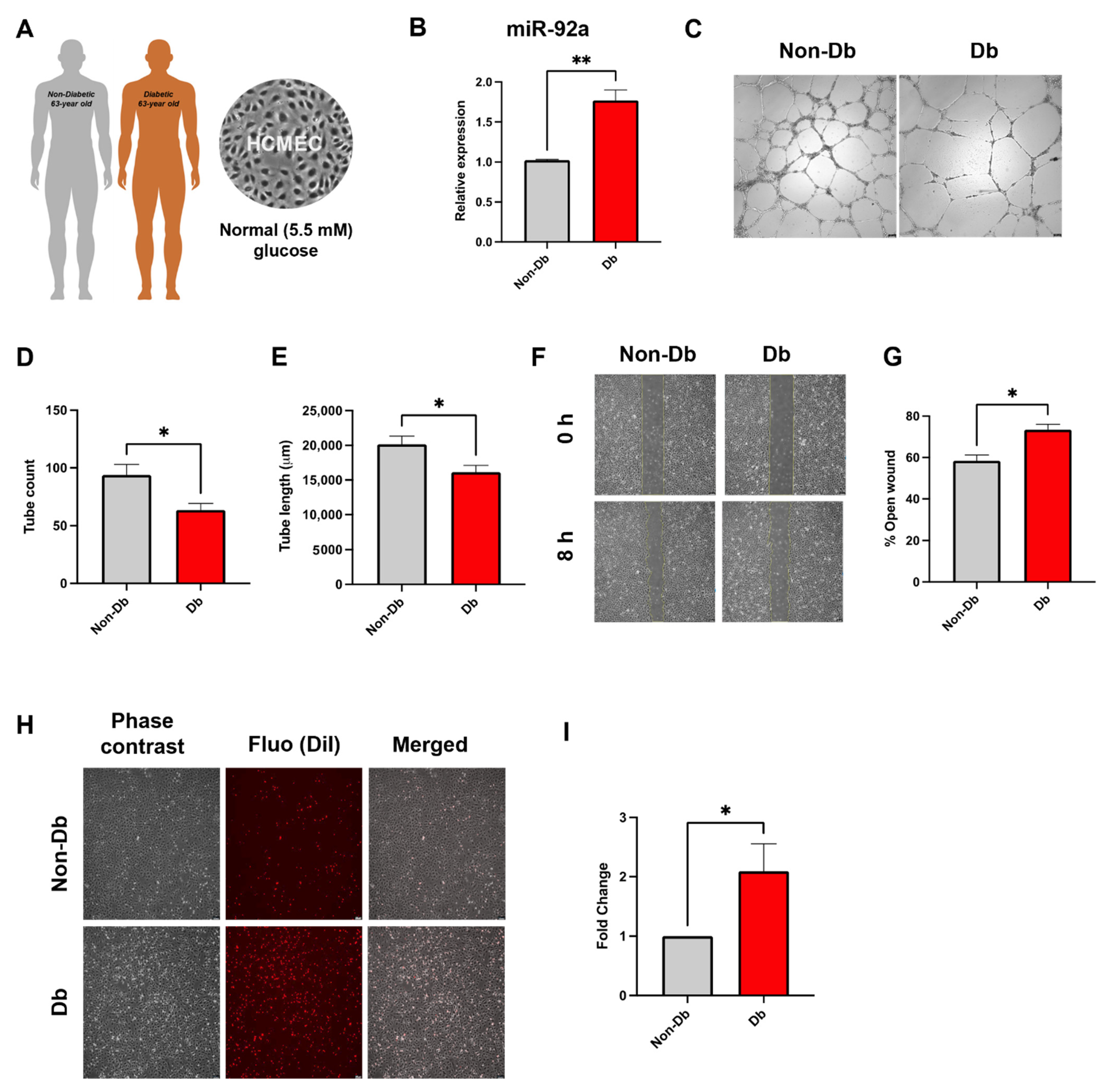
3.2. Inflammatory Phenotype of Diabetic HCMEC
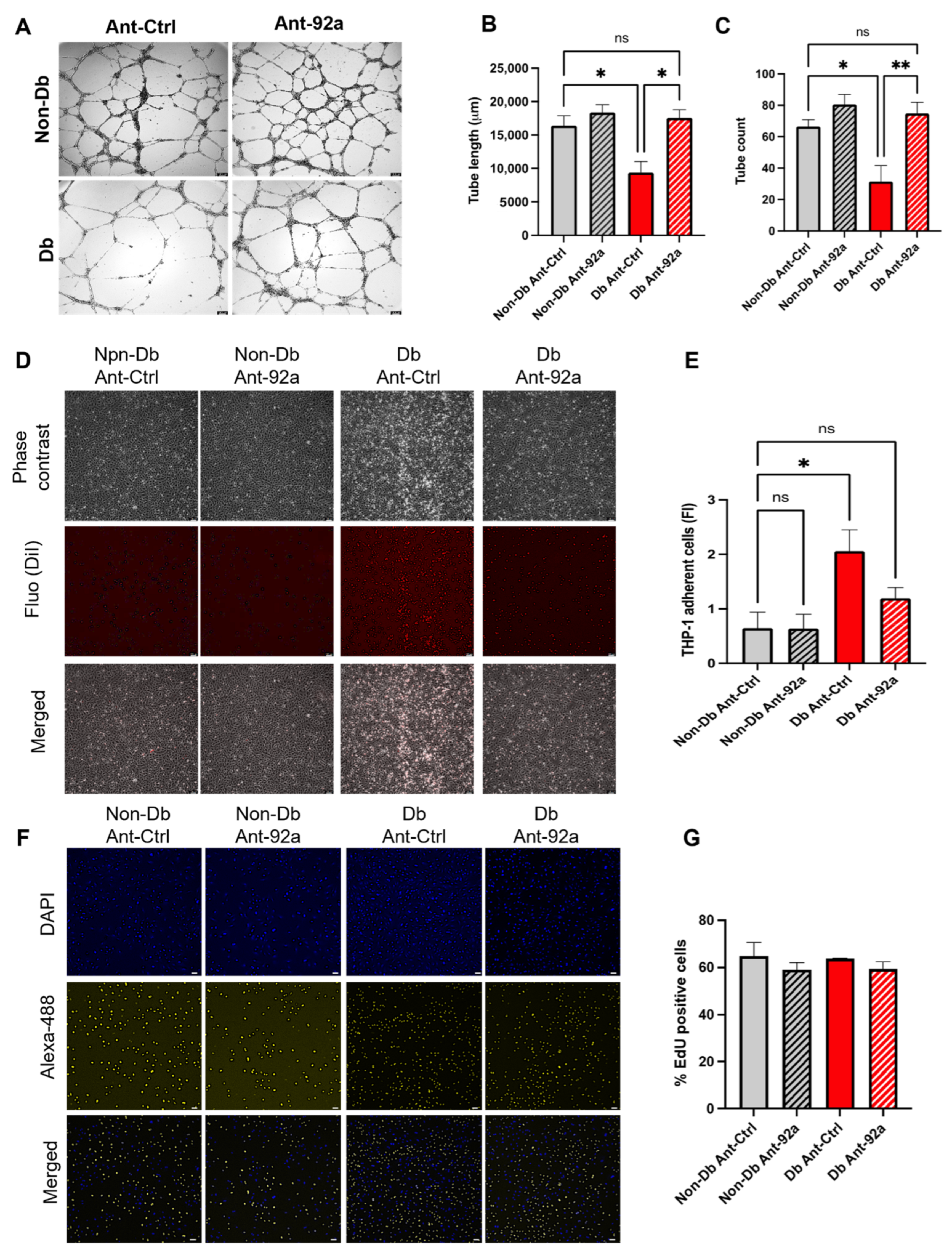
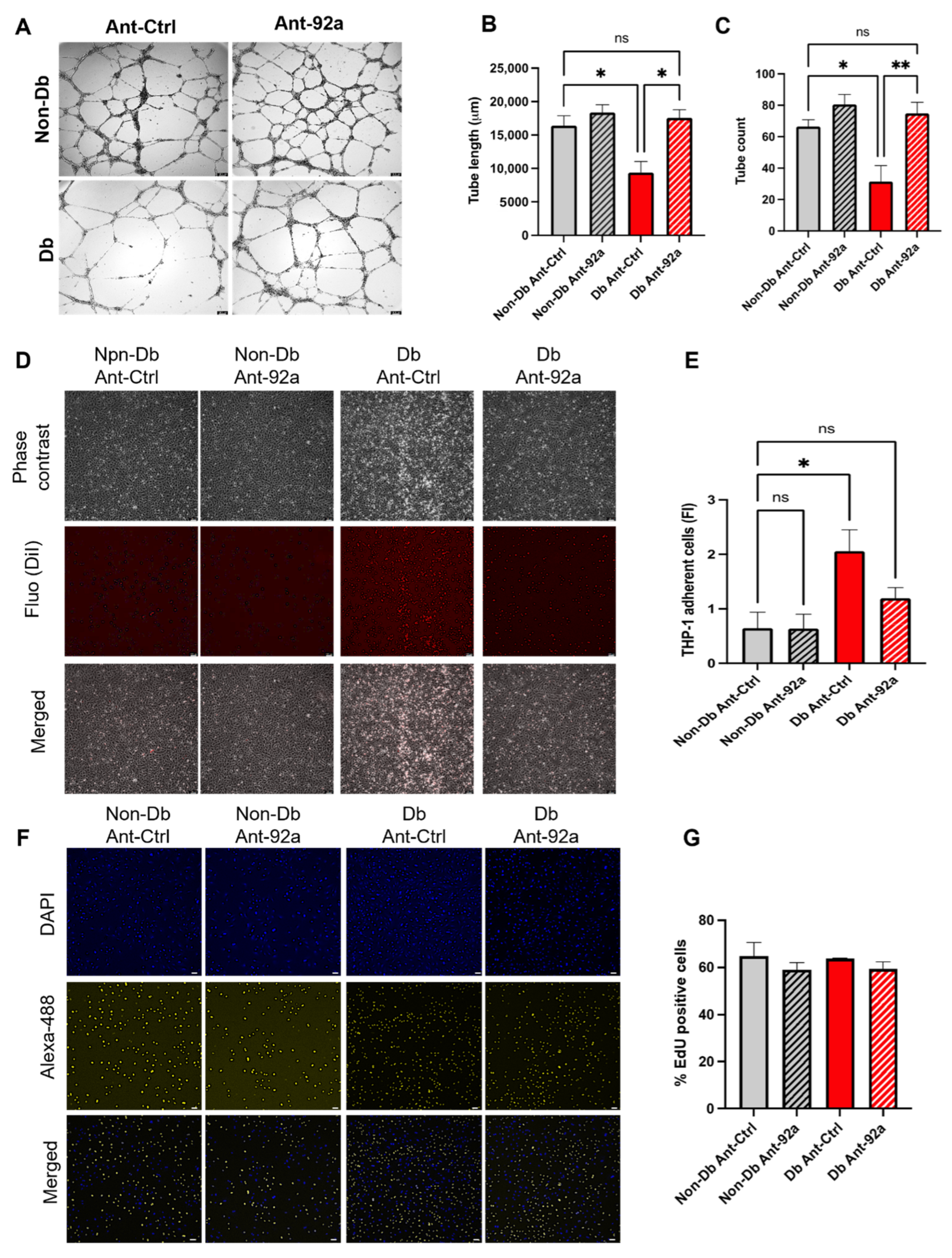
3.3. Downregulation of miR-92a Reverses Diabetic Phenotype
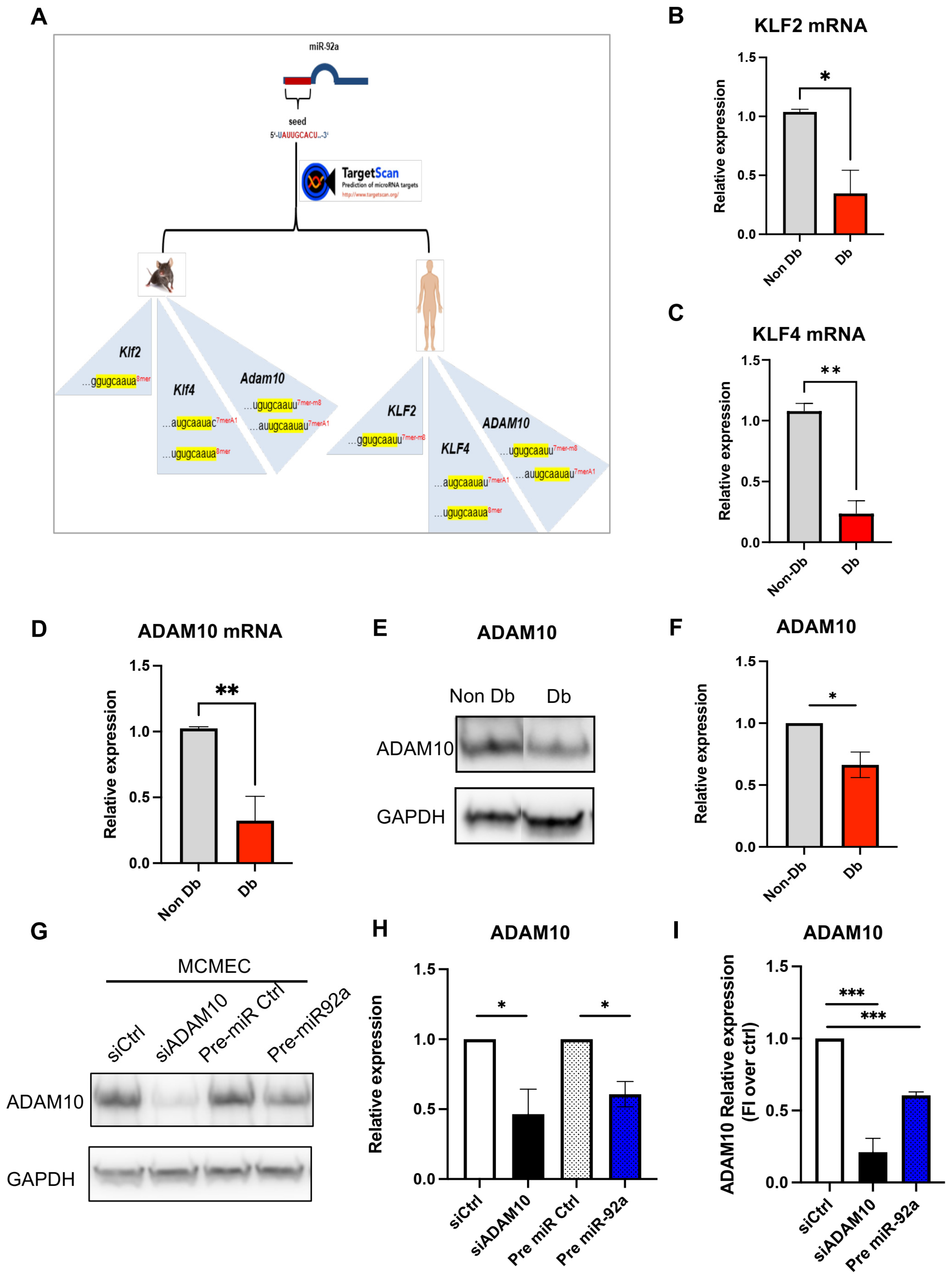
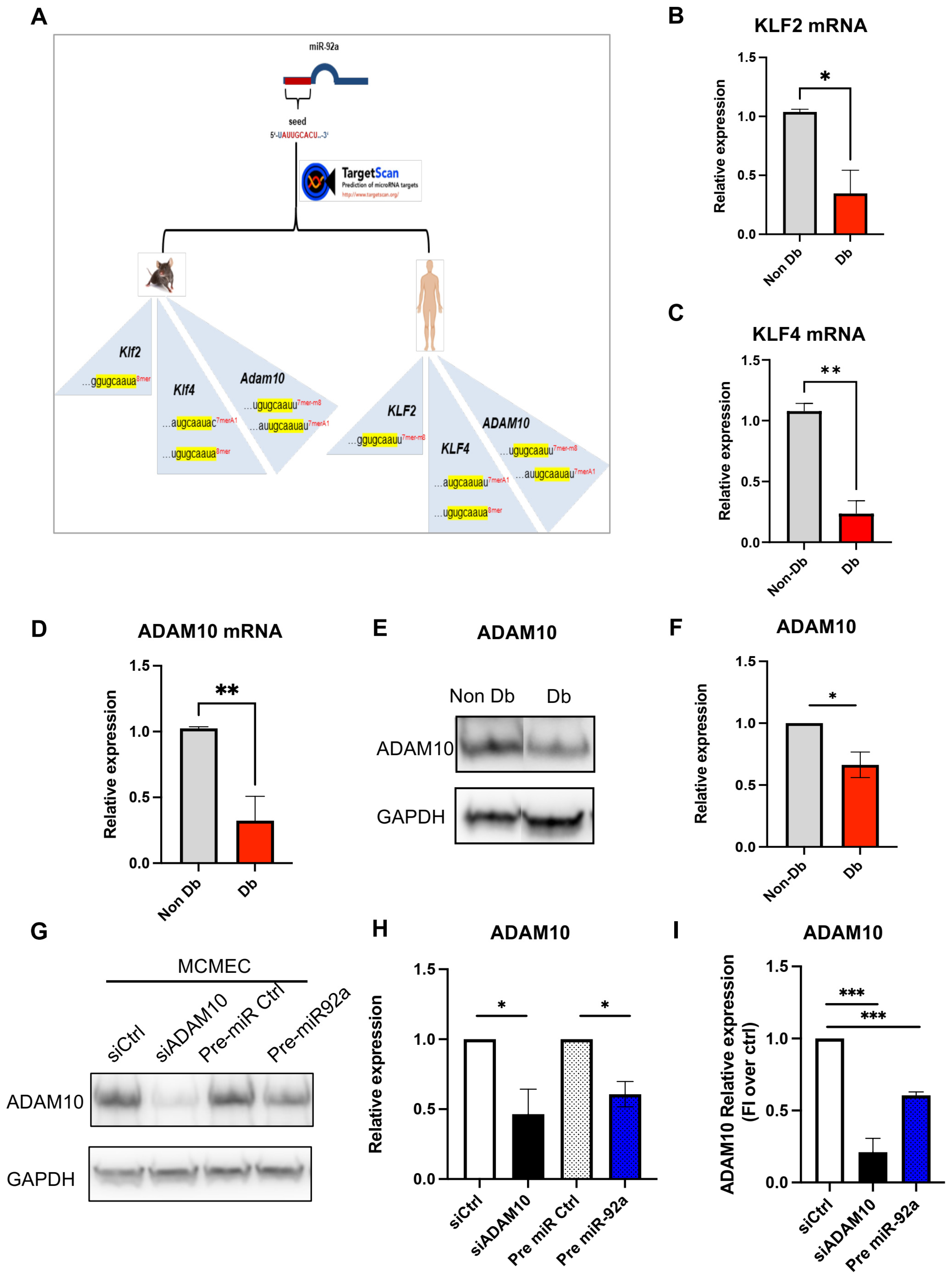
3.4. MiR-92a Downstream Targets
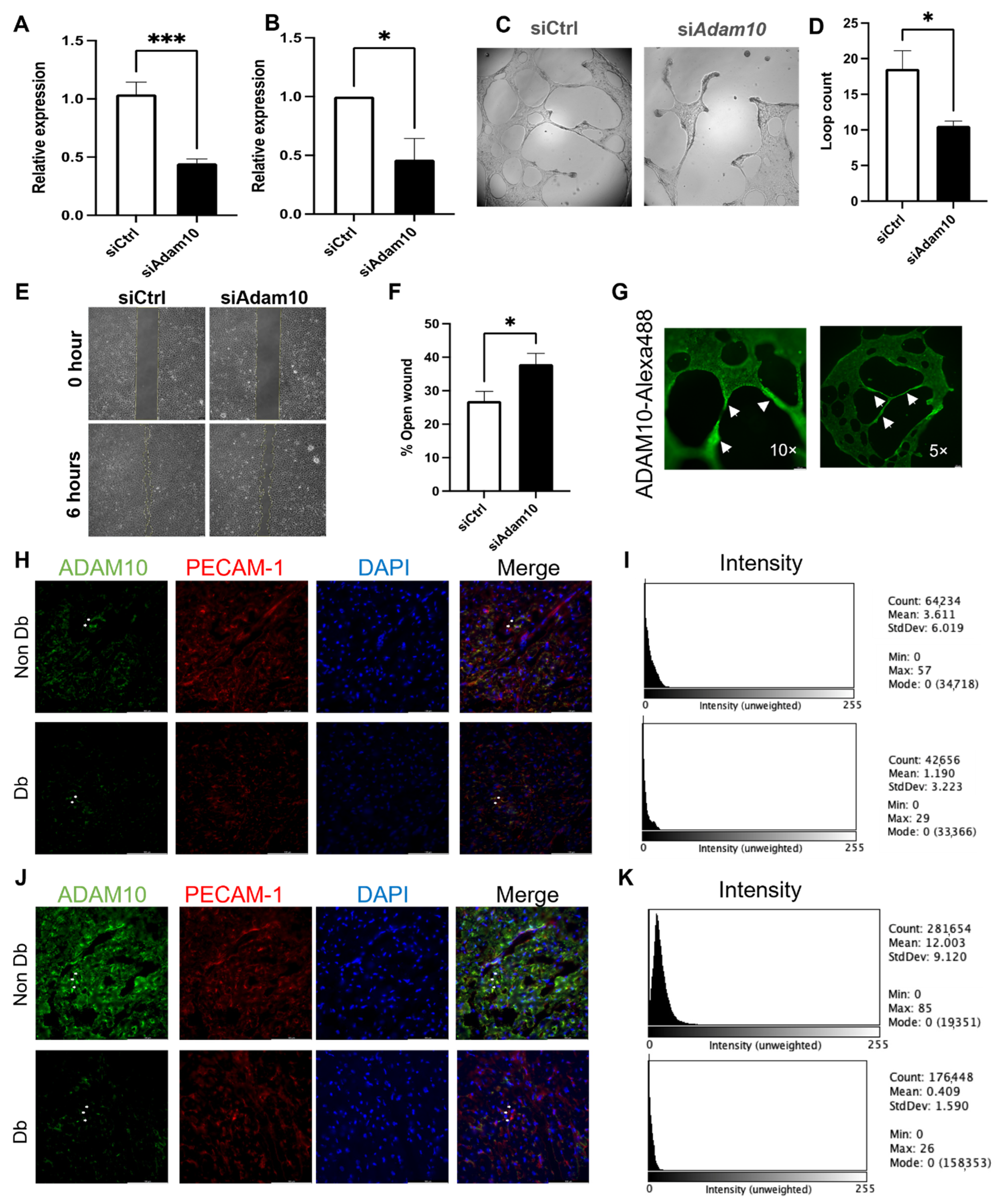
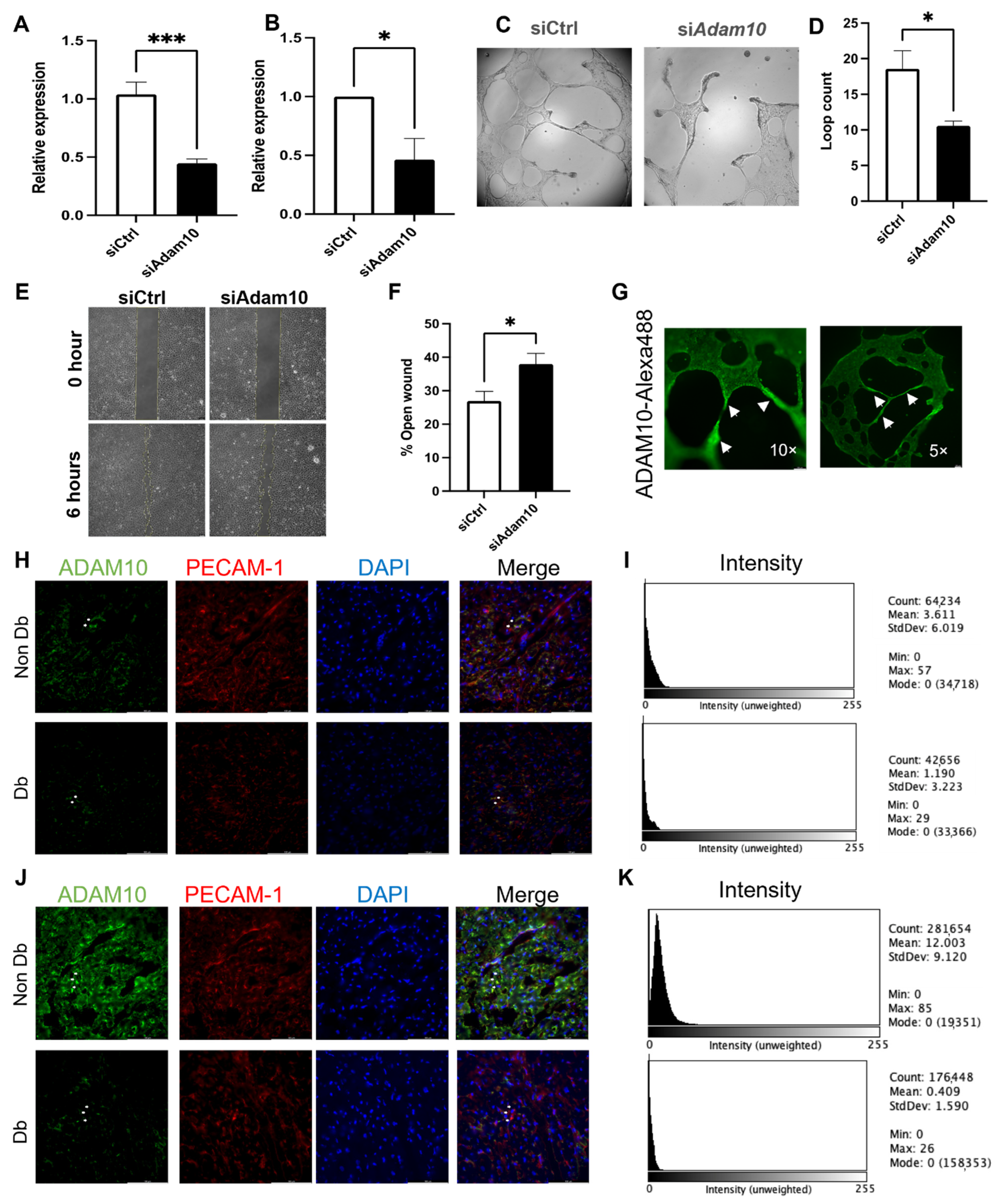
3.5. ADAM10 Is Important for Microvascular Endothelial Behavior
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar]
- Hinkel, R.; Howe, A.; Renner, S.; Ng, J.; Lee, S.; Klett, K.; Kaczmarek, V.; Moretti, A.; Laugwitz, K.L.; Skroblin, P.; et al. Diabetes Mellitus-Induced Microvascular Destabilization in the Myocardium. J. Am. Coll. Cardiol. 2017, 69, 131–143. [Google Scholar] [CrossRef]
- Bolognese, L.; Carrabba, N.; Parodi, G.; Santoro, G.M.; Buonamici, P.; Cerisano, G.; Antoniucci, D. Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation 2004, 109, 1121–1126. [Google Scholar] [CrossRef]
- Tate, M.; Grieve, D.J.; Ritchie, R.H. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin. Sci. 2017, 131, 897–915. [Google Scholar] [CrossRef]
- Pechlivani, N.; Ajjan, R.A. Thrombosis and Vascular Inflammation in Diabetes: Mechanisms and Potential Therapeutic Targets. Front. Cardiovasc. Med. 2018, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Hinkel, R.; Penzkofer, D.; Zuhlke, S.; Fischer, A.; Husada, W.; Xu, Q.F.; Baloch, E.; van Rooij, E.; Zeiher, A.M.; Kupatt, C.; et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation 2013, 128, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabi, R.O.; Farber, G.; Blobel, C.P. Intriguing Roles for Endothelial ADAM10/Notch Signaling in the Development of Organ-Specific Vascular Beds. Physiol. Rev. 2018, 98, 2025–2061. [Google Scholar] [CrossRef] [PubMed]
- Farber, G.; Parks, M.M.; Lustgarten Guahmich, N.; Zhang, Y.; Monette, S.; Blanchard, S.C.; Di Lorenzo, A.; Blobel, C.P. ADAM10 controls the differentiation of the coronary arterial endothelium. Angiogenesis 2019, 22, 237–250. [Google Scholar] [CrossRef]
- Song, W.; Zhang, C.L.; Gou, L.; He, L.; Gong, Y.Y.; Qu, D.; Zhao, L.; Jin, N.; Chan, T.F.; Wang, L.; et al. Endothelial TFEB (Transcription Factor EB) Restrains IKK (IκB Kinase)-p65 Pathway to Attenuate Vascular Inflammation in Diabetic db/db Mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radmanesh, F.; Sadeghi Abandansari, H.; Ghanian, M.H.; Pahlavan, S.; Varzideh, F.; Yakhkeshi, S.; Alikhani, M.; Moradi, S.; Braun, T.; Baharvand, H. Hydrogel-mediated delivery of microRNA-92a inhibitor polyplex nanoparticles induces localized angiogenesis. Angiogenesis 2021, 24, 657–676. [Google Scholar] [CrossRef]
- Morris, G.; Puri, B.K.; Olive, L.; Carvalho, A.; Berk, M.; Walder, K.; Gustad, L.T.; Maes, M. Endothelial dysfunction in neuroprogressive disorders-causes and suggested treatments. BMC Med. 2020, 18, 305. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Xu, S.; Xu, Y.; Little, P.J.; Jin, Z.G. Targeting Mechanosensitive Transcription Factors in Atherosclerosis. Trends Pharmacol. Sci. 2019, 40, 253–266. [Google Scholar] [CrossRef]
- Stachel, G.; Trenkwalder, T.; Götz, F.; El Aouni, C.; Muenchmeier, N.; Pfosser, A.; Nussbaum, C.; Sperandio, M.; Hatzopoulos, A.K.; Hinkel, R.; et al. SDF-1 fused to a fractalkine stalk and a GPI anchor enables functional neovascularization. Stem Cells 2013, 31, 1795–1805. [Google Scholar] [CrossRef]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, X.; Icli, B.; Feinberg, M.W. Emerging Roles for MicroRNAs in Diabetic Microvascular Disease: Novel Targets for Therapy. Endocr. Rev. 2017, 38, 145–168. [Google Scholar] [CrossRef] [Green Version]
- Solly, E.L.; Psaltis, P.J.; Bursill, C.A.; Tan, J.T.M. The Role of miR-181c in Mechanisms of Diabetes-Impaired Angiogenesis: An Emerging Therapeutic Target for Diabetic Vascular Complications. Front. Pharmacol. 2021, 12, 718679. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liu, Z.; Zhou, L. Roles of miR-17-92 Cluster in Cardiovascular Development and Common Diseases. BioMed Res. Int. 2017, 2017, 9102909. [Google Scholar] [CrossRef] [Green Version]
- Landskroner-Eiger, S.; Qiu, C.; Perrotta, P.; Siragusa, M.; Lee, M.Y.; Ulrich, V.; Luciano, A.K.; Zhuang, Z.W.; Corti, F.; Simons, M.; et al. Endothelial miR-17∼92 cluster negatively regulates arteriogenesis via miRNA-19 repression of WNT signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 12812–12817. [Google Scholar] [CrossRef] [Green Version]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef]
- Naito, H.; Iba, T.; Takakura, N. Mechanisms of new blood-vessel formation and proliferative heterogeneity of endothelial cells. Int. Immunol. 2020, 32, 295–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assar, M.E.; Angulo, J.; Rodríguez-Mañas, L. Diabetes and ageing-induced vascular inflammation. J. Physiol. 2016, 594, 2125–2146. [Google Scholar] [CrossRef] [Green Version]
- Pollack, R.M.; Donath, M.Y.; LeRoith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39 (Suppl. 2), S244–S252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.F.; Wu, K.J. Endothelial Transdifferentiation of Tumor Cells Triggered by the Twist1-Jagged1-KLF4 Axis: Relationship between Cancer Stemness and Angiogenesis. Stem Cells Int. 2016, 2016, 6439864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, A.T.; Tian, H.; Anih, E.; Recio, F.O., 3rd; Shatat, M.A.; Johnson, T.; Liao, X.; Ramirez-Bergeron, D.L.; Proweller, A.; Ishikawa, M.; et al. Endothelial Kruppel-like factor 4 regulates angiogenesis and the Notch signaling pathway. J. Biol. Chem. 2014, 289, 12016–12028. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.; Bonauer, A.; Dimmeler, S. RNA Therapeutics in Cardiovascular Disease. Circ. Res. 2018, 123, 205–220. [Google Scholar] [CrossRef]
- Abplanalp, W.T.; Fischer, A.; John, D.; Zeiher, A.M.; Gosgnach, W.; Darville, H.; Montgomery, R.; Pestano, L.; Allée, G.; Paty, I.; et al. Efficiency and Target Derepression of Anti-miR-92a: Results of a First in Human Study. Nucleic Acid 2020, 30, 335–345. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target mRNA | Match Position | Site Type | Site Context Score |
|---|---|---|---|
| Hs-ADAM10 | 490–496 | 7mer-m8 | −0.22 |
| 510–516 | 7mer-A1 | −0.13 | |
| Hs-KLF2 | 242–249 | 8mer | −0.50 |
| Hs-KLF4 | 362–368 | 7mer-A1 | −0.06 |
| 674–681 | 8mer | −0.41 | |
| Mm-Adam10 | 489–495 | 7mer-m8 | −0.22 |
| 509–515 | 7mer-A1 | −0.10 | |
| Mm-Klf2 | 214–221 | 8mer | −0.57 |
| Mm-Klf4 | 433–439 | 7mer-A1 | −0.10 |
| 751–758 | 8mer | −0.36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samak, M.; Kaltenborn, D.; Kues, A.; Le Noble, F.; Hinkel, R.; Germena, G. Micro-RNA 92a as a Therapeutic Target for Cardiac Microvascular Dysfunction in Diabetes. Biomedicines 2022, 10, 58. https://doi.org/10.3390/biomedicines10010058
Samak M, Kaltenborn D, Kues A, Le Noble F, Hinkel R, Germena G. Micro-RNA 92a as a Therapeutic Target for Cardiac Microvascular Dysfunction in Diabetes. Biomedicines. 2022; 10(1):58. https://doi.org/10.3390/biomedicines10010058
Chicago/Turabian StyleSamak, Mostafa, Diana Kaltenborn, Andreas Kues, Ferdinand Le Noble, Rabea Hinkel, and Giulia Germena. 2022. "Micro-RNA 92a as a Therapeutic Target for Cardiac Microvascular Dysfunction in Diabetes" Biomedicines 10, no. 1: 58. https://doi.org/10.3390/biomedicines10010058
APA StyleSamak, M., Kaltenborn, D., Kues, A., Le Noble, F., Hinkel, R., & Germena, G. (2022). Micro-RNA 92a as a Therapeutic Target for Cardiac Microvascular Dysfunction in Diabetes. Biomedicines, 10(1), 58. https://doi.org/10.3390/biomedicines10010058