
Co-Entrapment of Sorafenib and Cisplatin Drugs and iRGD Tumour Homing Peptide by Poly[ε-caprolactone-co-(12-hydroxystearate)] Copolymer

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
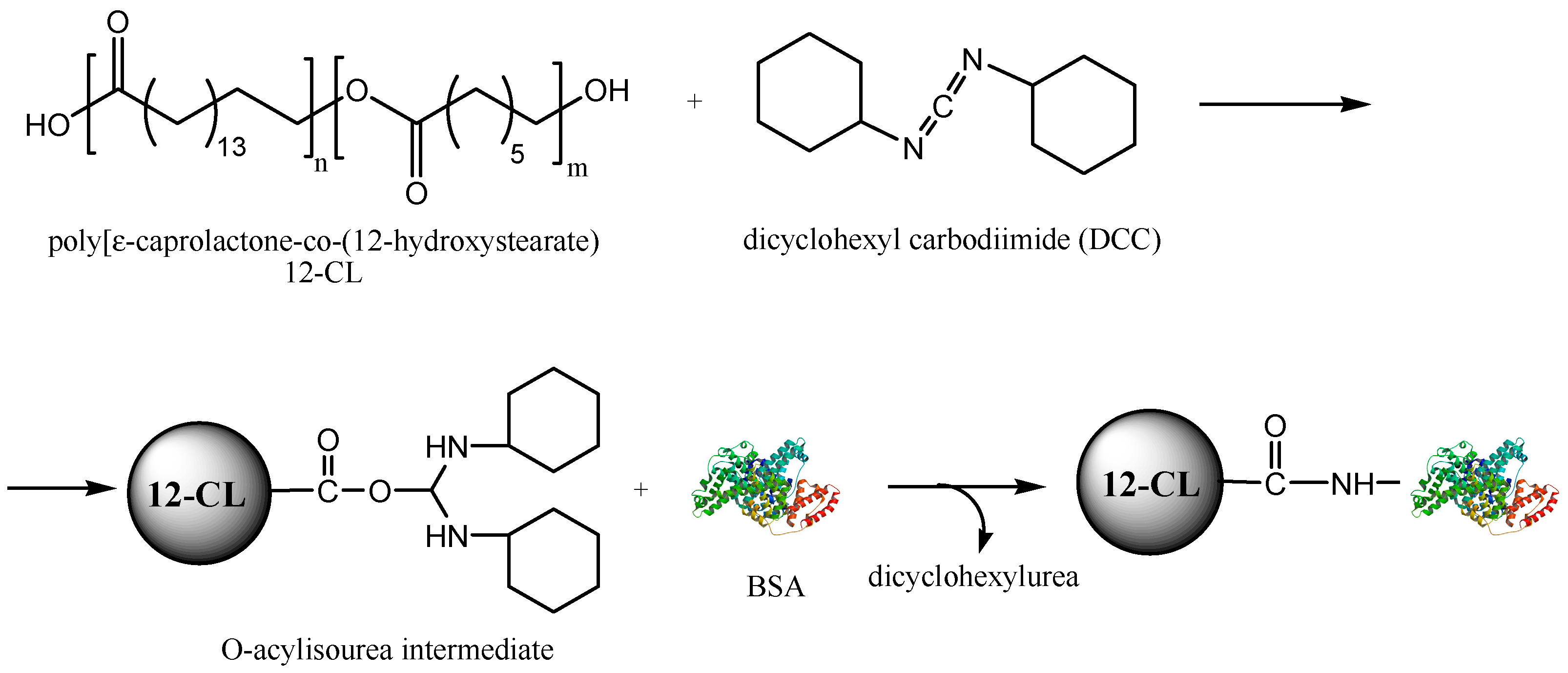
2.2. Biocatalytic Synthesis and Characterisation of the ε-Caprolactone-12-hydroxystearic Acid Copolymer
2.3. Preparation of Nanoparticles by Double Emulsion Solvent Evaporation Method
2.4. Characterisation of Nanoparticles
2.4.1. Particle Size, Morphology and Zeta Potential Analysis
2.4.2. Yield and Encapsulation Efficiency
2.4.3. Quantification of BSA by Micro Bicinchoninic Acid Protein Assay
2.4.4. In Vitro Drug Release
2.4.5. In Vitro Cytotoxicity Study
2.4.6. Data Processing
3. Results and Discussion
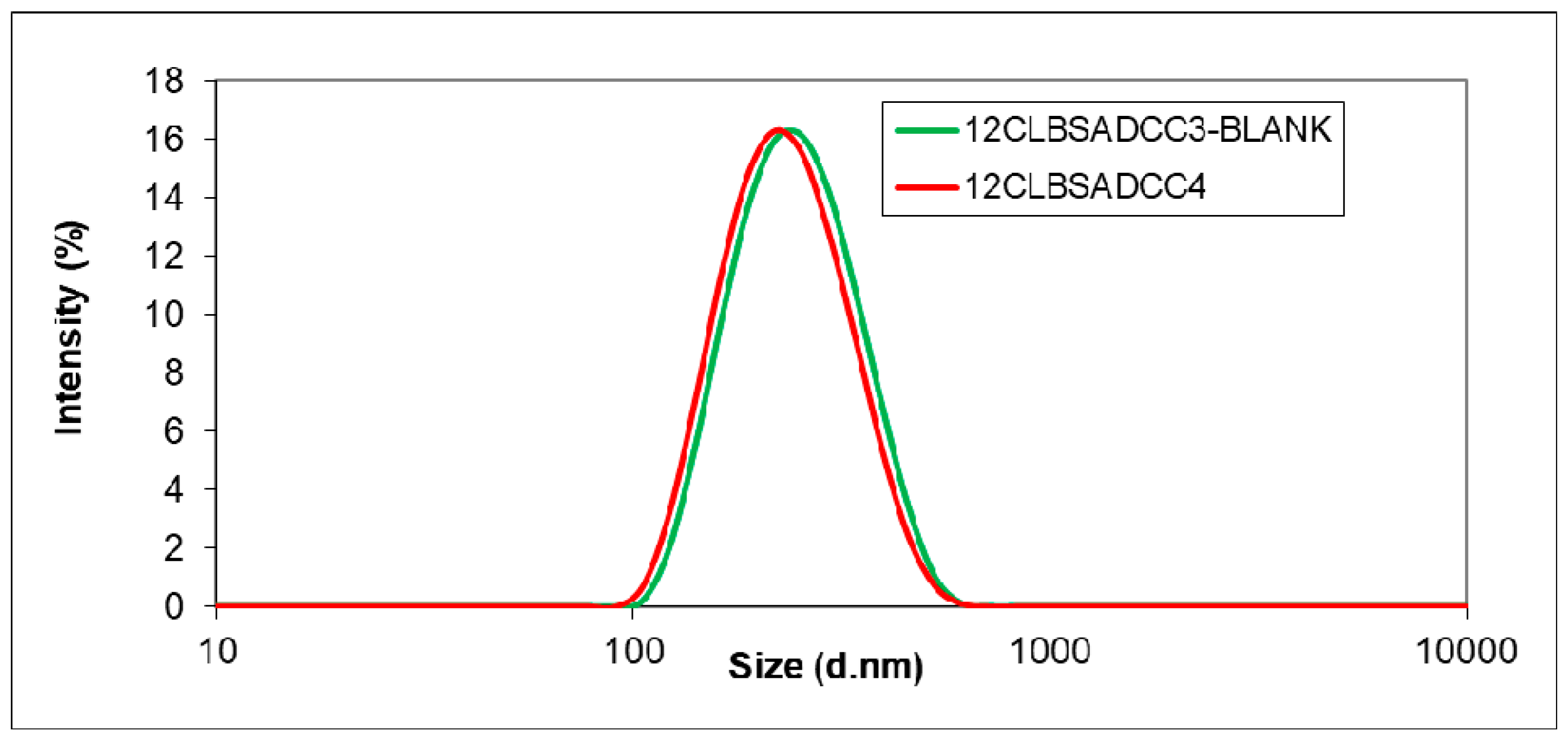
3.1. Encapsulation of the Active Agents using BSA as Additive
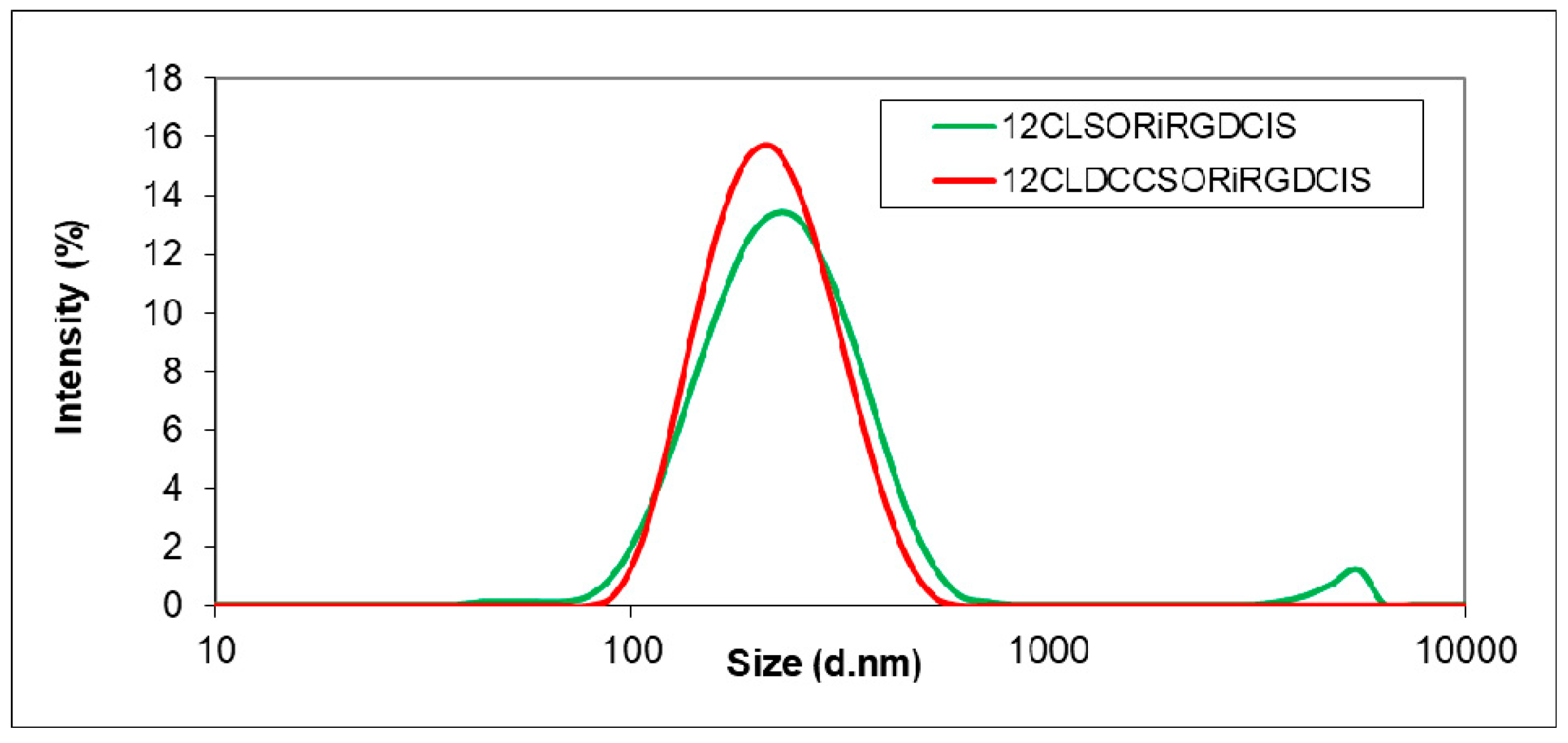
3.2. Simultaneous Encapsulation of Sorafenib and Cisplatin using iRGD
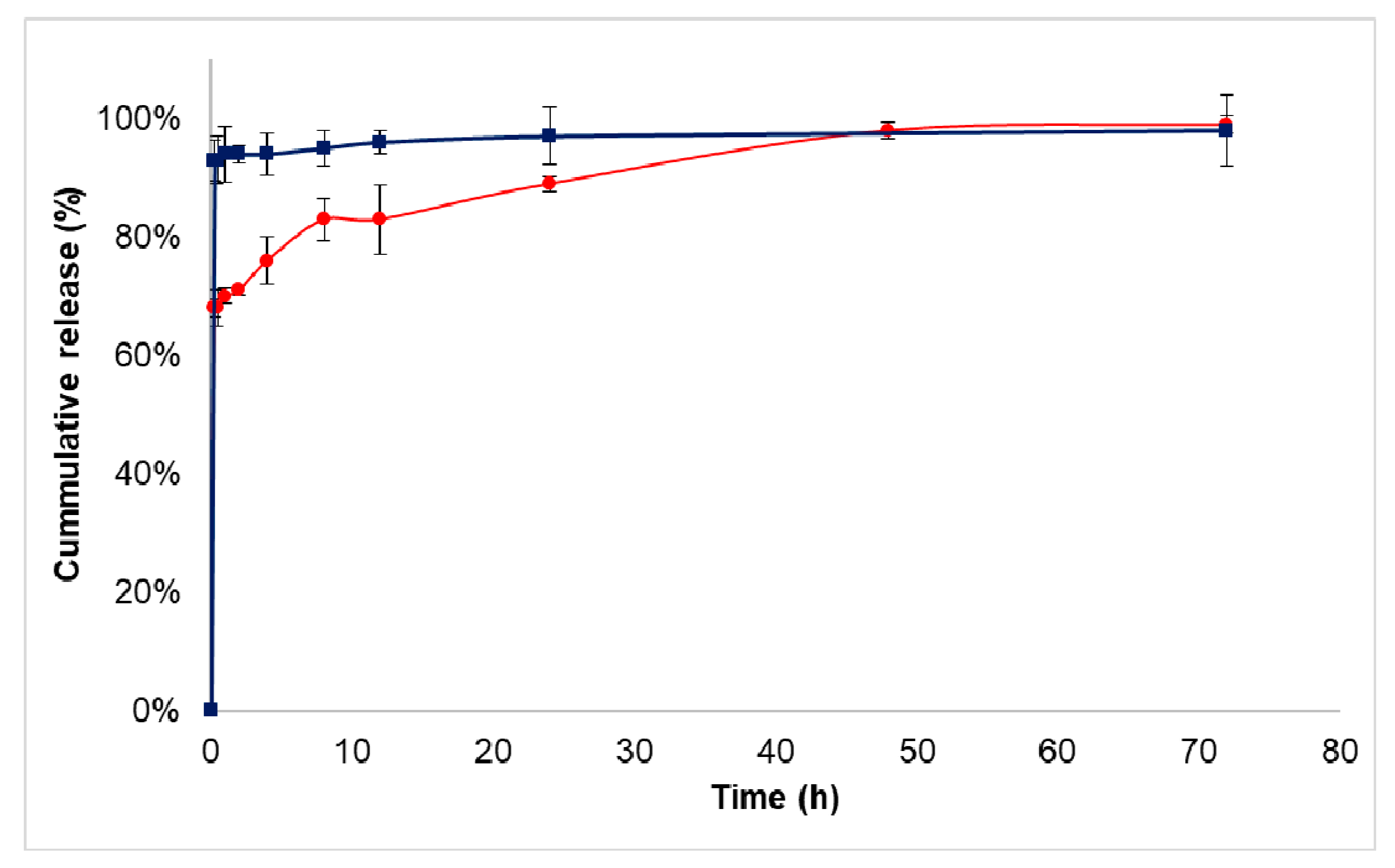
3.3. Drug Release Study
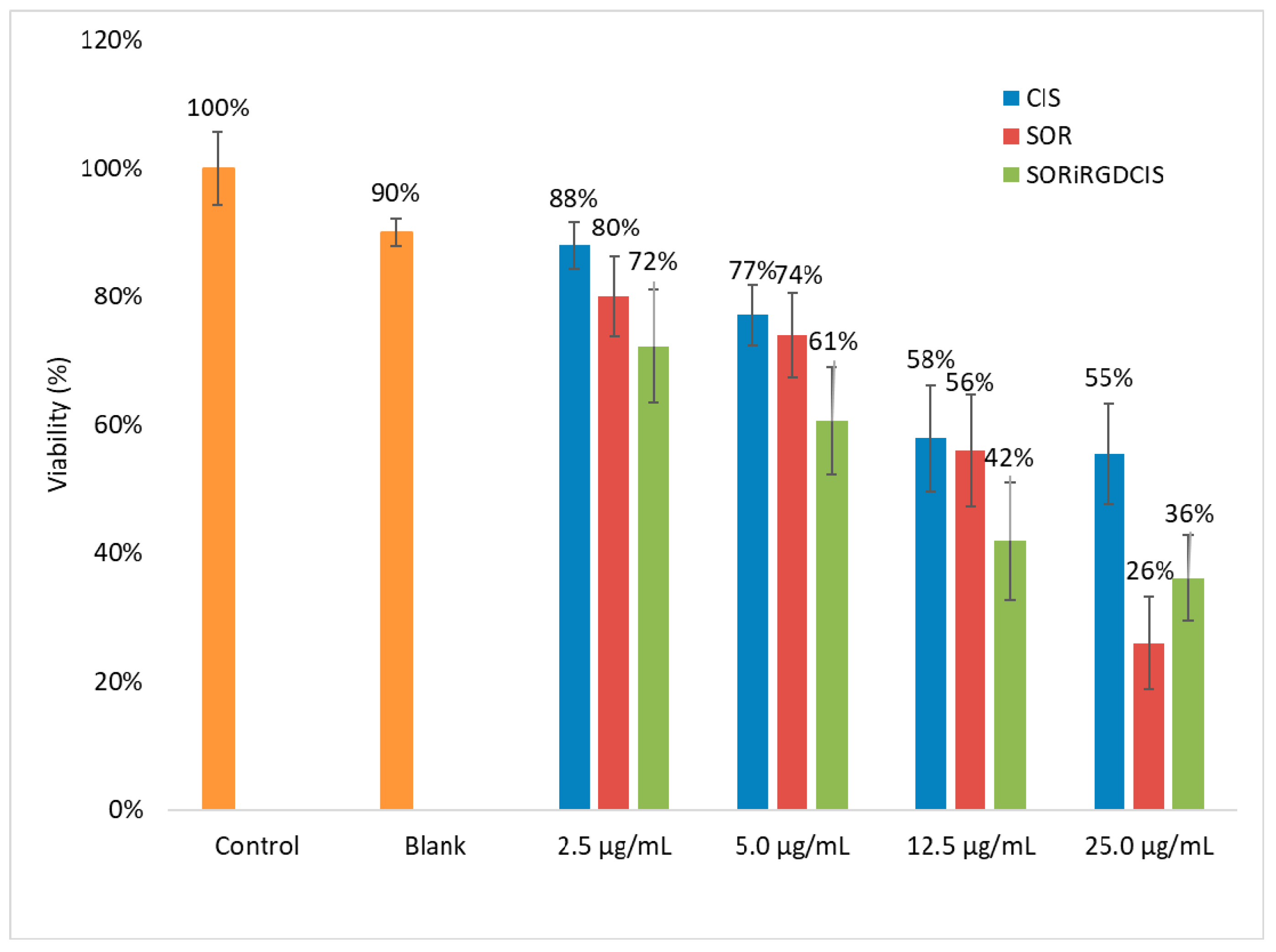
3.4. Cytotoxicity Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Kim, G.J.; Nie, S.; Shin, D.M. Nanotechnology in cancer therapeutics: Bioconjugated nanoparticles for drug delivery. Mol. Cancer Ther. 2006, 5, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, D.R.; Tangen, K.M.; Sadeh, M.; Seksenyan, A.; Neisewander, B.L.; Mehta, A.I.; Linninger, A.A. Systems engineers’ role in biomedical research. Convection-enhanced drug delivery. In Computer Aided Chemical Engineering; Manca, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 42, pp. 271–302. [Google Scholar] [CrossRef]
- Seo, S.-J.; Chen, M.; Wang, H.; Kang, M.S.; Leong, K.W.; Kim, H.-W. Extra- and intra-cellular fate of nanocarriers under dynamic interactions with biology. Nano Today 2017, 14, 84–99. [Google Scholar] [CrossRef]
- Feczkó, T.; Piiper, A.; Pleli, T.; Schmithals, C.; Denk, D.; Hehlgans, S.; Rödel, F.; Vogl, T.J.; Wacker, M.G. Theranostic Sorafenib-Loaded Polymeric Nanocarriers Manufactured by Enhanced Gadolinium Conjugation Techniques. Pharmaceutics 2019, 11, 489. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Peer, D.; Langer, R. Reshaping the future of nanopharmaceuticals: Ad Iudicium. ACS Nano 2011, 5, 8454–8458. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.T.; Holland, N.B. Multifunctional nanoparticles for use in theranostic applications. Drug Deliv. Transl. Res. 2015, 5, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Li, Y. Sorafenib-Loaded Ligand-Functionalized Polymer-Lipid Hybrid Nanoparticles for Enhanced Therapeutic Effect Against Liver Cancer. J. Nanosci. Nanotechnol. 2019, 19, 6866–6871. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Gilabert, M.; Bruix, J.; Raoul, J.-L. Treatment of intermediate-stage hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2014, 11, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Gbolahan, O.B.; Schacht, M.A.; Beckley, E.W.; LaRoche, T.P.; O’Neil, B.H.; Pyko, M. Locoregional and systemic therapy for hepatocellular carcinoma. J. Gastrointest. Oncol. 2017, 8, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Santoro, A. Sorafenib: A Review of its Use in Advanced Hepatocellular Carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; He, C.; Kron, S.J.; Lin, W. Nanoparticle formulations of cisplatin for cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Khare, V.; Dubey, R.; Saneja, A.; Kushwaha, M.; Singh, G.; Sherma, N.; Chandan, B.; Gupta, P.N. Biodegradable polymeric system for cisplatin delivery: Development, in vitro characterization and investigation of toxicity profile. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 38, 85–93. [Google Scholar] [CrossRef]
- Gryparis, E.C.; Mattheolabakis, G.; Bikiaris, D.; Avgoustakis, K. Effect of Conditions of Preparation on the Size and Encapsulation Properties of PLGA-mPEG Nanoparticles of Cisplatin. Drug Deliv. 2007, 14, 371–380. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Sig. Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Kaur, I.P.; Singh, H. Nanostructured drug delivery for better management of tuberculosis. J. Control Release 2014, 184, 36–50. [Google Scholar] [CrossRef] [PubMed]
- ud Din, F.; Aman, W.; Ullah, I.; Qureshi, O.S.; Mustapha, O.; Shafique, S.; Zeb, A. Effective use of nanocarriers as drug delivery systems for the treatment of selected tumors. Int. J. Nanomed. 2017, 12, 7291–7309. [Google Scholar] [CrossRef]
- Martinez, N.Y.; Andrade, P.F.; Durán, N.; Cavalitto, S. Development of Double Emulsion Nanoparticles for the Encapsulation of Bovine Serum Albumin. Colloids Surf. B 2017, 158, 190–196. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.; Yang, X.; Zhang, S.; Cui, C. Preparation optimization of bovine serum albumin nanoparticles and its application for siRNA delivery. Drug Des. Dev. Ther. 2021, 15, 1531–1547. [Google Scholar] [CrossRef]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle system in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, J.; Fu, C.; Xie, X.; Peng, F.; You, J.; Tang, H.; Wang, Z.; Li, P.; Chen, J. iRGD-modified lipid-polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting ability. Int. J. Nanomed. 2017, 12, 4147–4162. [Google Scholar] [CrossRef]
- Zhong, Y.; Su, T.; Shi, Q.; Feng, Y.; Tao, Z.; Huang, Q.; Li, L.; Hu, L.; Li, S.; Tan, H.; et al. Co-Administration Of iRGD Enhances Tumor-Targeted Delivery and Anti-Tumor Effects of Paclitaxel-Loaded PLGA Nanoparticles for Colorectal Cancer Treatment. Int. J. Nanomed. 2019, 14, 8543–8560. [Google Scholar] [CrossRef]
- Todea, A.; Aparaschivei, D.; Badea, V.; Boeriu, C.G.; Peter, F. Biocatalytic route for the synthesis of oligoesters of hydroxy-fatty acids and ϵ-caprolactone. Biotechnol. J. 2018, 13, 1700629. [Google Scholar] [CrossRef]
- Kántor, I.; Aparaschivei, D.; Todea, A.; Biró, E.; Babos, G.; Szerényi, D.; Kakasi, B.; Péter, F.; Șișu, E.; Feczkó, T. Biocatalytic synthesis of poly[ε-caprolactone-co-(12-hydroxystearate)] copolymer for sorafenib nanoformulation useful in drug delivery. Catal. Today 2021, 366, 195–201. [Google Scholar] [CrossRef]
- Todea, A.; Dreavă, D.M.; Benea, I.C.; Bîtcan, I.; Peter, F.; Boeriu, C.G. Achievements and trends in biocatalytic synthesis of specialty polymers from biomass-derived monomers using lipases. Processes 2021, 9, 646. [Google Scholar] [CrossRef]
- Hevilla, V.; Sonseca, A.; Echeverría, C.; Muñoz-Bonilla, A.; Fernández-García, M. Enzymatic synthesis of polyesters and their bioapplications: Recent advances and perspectives. Macromol. Biosci. 2021, 21, 2100156. [Google Scholar] [CrossRef]
- Todea, A.; Otten, L.G.; Frissen, A.E.; Arends, I.W.C.E.; Peter, F.; Boeriu, C.G. Selectivity of lipases for estolides synthesis. Pure Appl. Chem. 2015, 87, 51. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, N.; Cui, M.; Liu, Z.; Liu, S. Investigation on the hydrolysis of the anticancer drug cisplatin by Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 2832–2836. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.; Amreddy, N.; Muralidharan, R.; Pathuri, G.; Gali, H.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. Chemodrug delivery using integrin-targeted PLGA-Chitosan nanoparticle for lung cancer therapy. Sci. Rep. 2017, 7, 14674. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Min, Y.; Rodgers, Z.; Man Au, K.; Hagan, C.T.; Zhang, M.; Roche, K.; Yang, F.; Wagner, K.; Wang, A.Z. Co-delivery of paclitaxel and cisplatin with biocompatible PLGA-PEG nanoparticles enhances chemoradiotherapy in non-small cell lung cancer models. J. Mater. Chem. B 2017, 5, 6049–6057. [Google Scholar] [CrossRef] [PubMed]
- Callari, M.; Aldrich-Wright, J.R.; de Souza, P.L.; Stenzel, M.H. Polymers with platinum drugs and other macromolecular metal complexes for cancer treatment. Prog. Polym. Sci. 2014, 39, 1614–1643. [Google Scholar] [CrossRef]
- Moreno, D.; Tros de Ilarduya, C.; Bandrés, A.; Buñuales, M.; Azcona, M.; García-Foncillas, J.; Garrido, M.J. Characterization of cisplatin cytotoxicity delivered from PLGA-system. Eur. J. Pharm. Biopharm. 2008, 68, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, A.; Darr, A. Controlled release of cisplatin and cancer cell apoptosis with cisplatin encapsulated poly(lactic-co-glycolic acid) nanoparticles. J. Biomed. Sci. Eng. 2013, 6, 586–592. [Google Scholar] [CrossRef][Green Version]
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Karydas, A.G.; Ithakissios, D.S. PLGA-mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control Release 2002, 79, 123–135. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | o/w Ratio | Polymer [%] | PVA [%] | BSA [mg] | EDC [mg] | Z-avg [nm] | PDI |
|---|---|---|---|---|---|---|---|
| 12CLBSA1 | 1/2 | 1 | 0.5 | 1 | 0 | 476.6 ± 9.3 | 0.450 ± 0.018 |
| 12CLBSA2 | 1/3 | 1 | 1 | 1 | 0 | 241.3 ± 5.8 | 0.341 ± 0.004 |
| 12CLBSA3 | 1/3 | 2 | 1 | 1 | 0 | 481.9 ± 39.6 | 0.692 ± 0.022 |
| 12CLBSA4 | 1/3 | 1 | 1 | 0.5 | 0 | 232.3 ± 10.2 | 0.360 ± 0.051 |
| 12CLBSA5 | 1/5 | 1 | 1 | 1 | 0 | 269.9 ± 7.8 | 0.355 ± 0.019 |
| 12CLBSA6 | 1/5 | 1 | 1 | 0.5 | 0 | 245.5 ± 3.1 | 0.275 ± 0.007 |
| 12CLBSA7 | 1/4 | 1 | 1 | 1 | 0 | 301.2 ± 7.6 | 0.461 ± 0.018 |
| 12CLBSA8 | 1/4 | 1 | 1 | 0.5 | 0 | 338.8 ± 11.6 | 0.568 ± 0.035 |
| 12CLBSA9 | 1/5 | 1 | 1 | 1 | 1 | 438.5 ± 25.1 | 0.810 ± 0.121 |
| 12CLBSA10 | 1/5 | 1 | 1 | 0.5 | 1 | 250.7 ± 8.3 | 0.353 ± 0.038 |
| 12CLBlank | 1/5 | 1 | 1 | 0 | 0 | 215.2 ± 1.4 | 0.222 ± 0.011 |
| Sample | Sorafenib [mg] | Cisplatin [mg] | EDC [mg] | Z-avg [nm] | PDI |
|---|---|---|---|---|---|
| 12CLBSASOR1 | 1 | 0 | 0 | 223.4 ± 2.4 | 0.243 ± 0.008 |
| 12CLBSASOR2 a | 1 | 0 | 1 | 274.4 ± 8.6 | 0.582 ± 0.018 |
| 12CLBSASOR3 b | 1 | 0 | 1 | 212.3 ± 0.8 | 0.258 ± 0.028 |
| 12CLBSASORCIS1 | 0.5 | 0.5 | 0 | 239.6 ± 3.7 | 0.378 ± 0.014 |
| 12CLBSASORCIS2 a | 0.5 | 0.5 | 1 | 196.1 ± 1.7 | 0.081 ± 0.013 |
| 12CLBSASORCIS3 b | 0.5 | 0.5 | 1 | 249.3 ± 8.1 | 0.362 ± 0.021 |
| 12CLBSACIS1 | 0 | 1 | 0 | 252.2 ± 15.0 | 0.413 ± 0.02 |
| 12CLBSACIS2 a | 0 | 1 | 1 | 246.3 ± 27.2 | 0.397 ± 0.070 |
| 12CLBSACIS3 b | 0 | 1 | 1 | 188.9 ± 2.0 | 0.091 ± 0.008 |
| Sample | BSA [mg] | DCC [mg] | Z-avg [nm] | PDI | EE BSA [%] | Yield [%] |
|---|---|---|---|---|---|---|
| 12CLBSADCC1 | 0.5 | 1 | 217.6 ± 2.0 | 0.129 ± 0.014 | 71 | - |
| 12CLBSADCC2 | 0.5 | 2 | 225.9 ± 2.2 | 0.115 ± 0.015 | 73 | 46 |
| 12CLBSADCC3 | 1 | 2 | 219.7 ± 1.7 | 0.121 ± 0.011 | 82 | 46 |
| 12CLBSADCCBlank | 0 | 2 | 233.4 ± 1.4 | 0.136 ± 0.020 | - | 47 |
| Sample | Cisplatin [mg] | DCC [mg] | Z-avg [nm] | PDI | Yield [%] | EE Cisplatin [%] |
|---|---|---|---|---|---|---|
| 12CLBSACISH4 | 0.5 | - | 204.6 ± 1.1 | 0.250 ± 0.014 | 40 | 24 |
| 12CLBSACISH5 Blank | - | 2 | 212.9 ± 2.4 | 0.171 ± 0.019 | 39 | - |
| 12CLBSACISH5 | 0.5 | 2 | 209.8 ± 3.0 | 0.209 ± 0.015 | 61 | 28 |
| Sample | iRGD [mg] | Z-avg [nm] | PDI | Zeta Potential [mV] | Yield [%] |
|---|---|---|---|---|---|
| DCCRGD Blank | - | 210.8 ± 1.6 | 0.119 ± 0.019 | −11.00 | 59 |
| DCCRGD1 | 0.5 | 223.7 ± 3.0 | 0.143 ± 0.03 | −11.20 | 49 |
| DCCRGD2 | 1 | 218.1 ± 0.9 | 0.158 ± 0.014 | −11.80 | 59 |
| Sample | Z-avg [nm] | PDI | Zeta Potential [mV] | Yield [%] | EE Sorafenib [%] | EE Cisplatin [%] | EE iRGD [%] |
|---|---|---|---|---|---|---|---|
| iRGDBlank | 220.6 ± 4.2 | 0.256 ± 0.016 | −9.42 | 63 ± 12 | - | - | - |
| SORiRGDCIS | 220.8 ± 2.5 | 0.221 ± 0.021 | −10.0 | 65 ± 11 | 54 ± 1.0 | 25 ± 1.0 | 42 ± 3.0 |
| DCCSOR-iRGDCIS | 205.9 ± 2.8 | 0.148 ± 0.012 | −11.8 | 74 ±13 | 55 ± 2.8 | 23 ± 2.1 | 29 ± 3.1 |
| Concentration Group | Normality Test (Shapiro–Wilk) | Homoscedasticity (Bartlett Test) | One-way-ANOVA/Kruskal–Wallis | Post-hoc (Tukey HSD/Nemenyi or Dunn) |
|---|---|---|---|---|
| 2.5 µg/mL | C: p = 0.440 S: p = 0.630 CS: p = 0.694 | p = 0.219 | F = 7.922, df = 2, p < 0.01 ** | C-CS: p < 0.01 ** |
| 5.0 µg/mL | C: p = 0.296 S: p = 0.374 CS: p = 0.027 | p = 0.158 | Chi-sq = 13.03, df = 2, p < 0.01 ** | C-CS: p < 0.01 ** S-CS: p < 0.05 * |
| 12.5 µg/mL | C: p = 0.041 S: p = 0.307 CS: p = 0.350 | p = 0.466 | Chi-sq = 14.25, df = 2, p < 0.001 *** | C-CS: p < 0.01 ** S-CS: p < 0.01 ** |
| 25.0 µg/mL | C: p = 0.048 S: p = 0.227 CS: p = 0.829 | p = 0.321 | Chi-sq = 18.12, df = 2, p < 0.001 *** | C-S: p < 0.05 * C-CS: p < 0.001 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kántor, I.; Dreavă, D.; Todea, A.; Péter, F.; May, Z.; Biró, E.; Babos, G.; Feczkó, T. Co-Entrapment of Sorafenib and Cisplatin Drugs and iRGD Tumour Homing Peptide by Poly[ε-caprolactone-co-(12-hydroxystearate)] Copolymer. Biomedicines 2022, 10, 43. https://doi.org/10.3390/biomedicines10010043
Kántor I, Dreavă D, Todea A, Péter F, May Z, Biró E, Babos G, Feczkó T. Co-Entrapment of Sorafenib and Cisplatin Drugs and iRGD Tumour Homing Peptide by Poly[ε-caprolactone-co-(12-hydroxystearate)] Copolymer. Biomedicines. 2022; 10(1):43. https://doi.org/10.3390/biomedicines10010043
Chicago/Turabian StyleKántor, Izolda, Diana Dreavă, Anamaria Todea, Francisc Péter, Zoltán May, Emese Biró, György Babos, and Tivadar Feczkó. 2022. "Co-Entrapment of Sorafenib and Cisplatin Drugs and iRGD Tumour Homing Peptide by Poly[ε-caprolactone-co-(12-hydroxystearate)] Copolymer" Biomedicines 10, no. 1: 43. https://doi.org/10.3390/biomedicines10010043
APA StyleKántor, I., Dreavă, D., Todea, A., Péter, F., May, Z., Biró, E., Babos, G., & Feczkó, T. (2022). Co-Entrapment of Sorafenib and Cisplatin Drugs and iRGD Tumour Homing Peptide by Poly[ε-caprolactone-co-(12-hydroxystearate)] Copolymer. Biomedicines, 10(1), 43. https://doi.org/10.3390/biomedicines10010043