Abstract
Lineage tracing studies have become a well-suited approach to reveal cellular hierarchies and tumor heterogeneity. Cellular heterogeneity, particularly in breast cancer, is still one of the main concerns regarding tumor progression and resistance to anti-cancer therapies. Here, we review the current knowledge about lineage tracing analyses that have contributed to an improved comprehension of the complexity of mammary tumors, highlighting how targeting different mammary epithelial cells and tracing their progeny can be useful to explore the intra- and inter-heterogeneity observed in breast cancer. In addition, we examine the strategies used to identify the cell of origin in different breast cancer subtypes and summarize how cellular plasticity plays an important role during tumorigenesis. Finally, we evaluate the clinical implications of lineage tracing studies and the challenges remaining to address tumor heterogeneity in breast cancer.
1. Introduction
Breast cancer is the most common cancer in women worldwide and is the second leading cause of cancer death in women, exceeded only by lung cancer (based on data from the World Health Organization, 2021). This type of cancer originates in the mammary gland, which is a ductal tree composed of two epithelial compartments: cells facing the ductal lumen called luminal cells (LCs), and basal cells (BCs) found in the outer layer with a capacity to contract, which includes basal progenitor cells and terminal differentiated myoepithelial cells. Luminal cells can be further subdivided into two independent subpopulations based on the expression of the hormone receptor, estrogen receptor alpha (ERα) [1] (Figure 1).
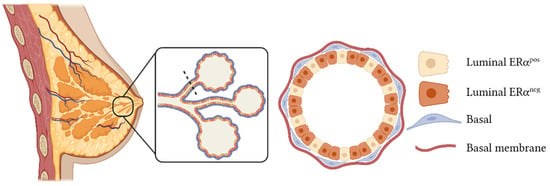
Figure 1.
Model of normal mammary gland structure. This tissue is composed of ducts, which are formed by three epithelial populations: basal cells, in contact with the basal membrane; estrogen receptor-positive (ERαpos) luminal cells; and estrogen receptor-negative (ERαneg) luminal cells. The dotted black line indicates the cross-section of the mammary duct represented in the magnified scheme on the right. This figure was created with Biorender.com (accessed on 12 December 2021).
In clinical practice, breast tumors are classified based on the histological expression of ERα, progesterone receptor (PR), receptor tyrosine-protein kinase ErbB-2 (HER2), and the proliferation marker Ki-67. They are divided into three main groups: hormone receptor-positive subtype, which includes tumors expressing ERα and/or PR, which are subclassified as luminal A or B depending on the percentage of Ki-67; HER2-positive tumors, defined by the presence of ERBB2/HER2 amplifications and loss of ERα expression; and triple-negative breast cancer (TNBC), characterized by the lack of expression of the aforementioned molecular markers [2] (Figure 2).
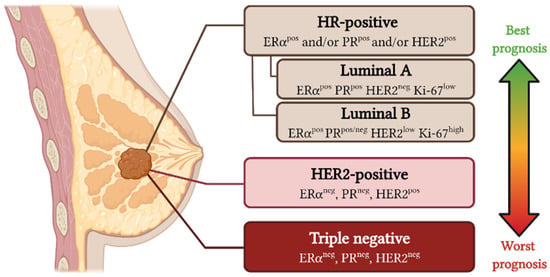
Figure 2.
Molecular classification of breast cancer subtypes. Breast cancer can be divided into three main subtypes depending on the histological expression of four markers (ERα, PR, HER2, and Ki-67): hormone receptor (HR)-positive, HER2-positive, and triple-negative. This figure was created with Biorender.com (accessed on 12 December 2021).
At the cellular level, breast cancer is a heterogeneous disease [3], thereby evoking disparate phenotypes not only between patients, but also within the tumor itself, known as inter- and intratumor heterogeneity, respectively [4]. It was suggested that this intratumor heterogeneity is perpetuated by tumor stem-like cells, leading many research groups to focus on the identification of biomarkers that allow them to target those cells responsible for tumor maintenance, including CD44 [5], CD29 and CD49f, [6], CD133 [7], Lgr5 [8], Procr [9], ALDH [10], or CD61 [11]. Although the origin of breast cancer, as many other types of cancer, remains largely elusive, a plausible theory is that adult mammary stem/progenitor cells, which are very long-lived compared with differentiated cells, are better targets for accumulating the multiple genetic mutations necessary for malignant transformation [12]; however, accumulating evidence has demonstrated that specific mutations in differentiated cells are also able to initiate a tumor [13,14,15,16]. Importantly, both luminal and basal cells are possible targets for malignant transformation [17,18].
During the last decade, lineage tracing studies have provided deeper insights into the cellular heterogeneity and molecular mechanisms underlying cellular plasticity in different subtypes of breast cancer. Accordingly, this review outlines the main milestones concerning this technology in the breast cancer context, as well as underlining unsolved questions and future prospects in the field.
2. Lineage Tracing Is the Gold-Standard Approach for Exploring Cellular Hierarchies and Tumor Heterogeneity in Breast Cancer
Genetic fate mapping studies commonly use a lineage-specific promoter followed by an inducible form of Cre recombinase fused to the mutant ligand-binding domain of the human estrogen receptor (CreER, CreERT, or CreERT2) [19,20,21], which does not bind to endogenous estradiol, but can be activated by the administration of synthetic ligands, such as tamoxifen or 4-hydroxytamoxifen (4-OHT). Furthermore, this Cre line needs to be mated with a reporter line carrying the β-galactosidase enzyme [22], one or several fluorescent protein(s) [23,24,25,26,27], or barcoding sequences [28,29], in order to monitor the targeted cells. In this way, tamoxifen or 4-OHT drive the inducible Cre to the nucleus, where it promotes the recombination of loxP sequences, allowing the expression of the reporter (Figure 3A). The use of this particular system has been controversial, since high doses of tamoxifen can delay mammary gland development [30]; however, the use of low doses (0.1 mg/g of mouse body weight) is a good balance between correct mammary epithelial development and sufficient labeling efficiency [31]. Alternatively, other studies have opted for tetracycline-inducible systems, where tetracycline administration, or its analogue doxycycline (Dox), enables the expression of a reverse tetracycline-controlled transactivator (rtTA). Activated rtTA will bind to tetracycline response elements (TRE), which trigger the expression of Cre recombinase to mediate the recombination of loxP sequences located at the reporter transgene [32,33] (Figure 3B). Advantageously, this system allows us to perform lineage tracing at saturation [34], relying on the long-term administration of Dox and inducing reporter recombination in every single cell of a given lineage, overcoming the low recombination efficiency obtained by classic lineage tracing experiments using ER-dependent Cre lines.
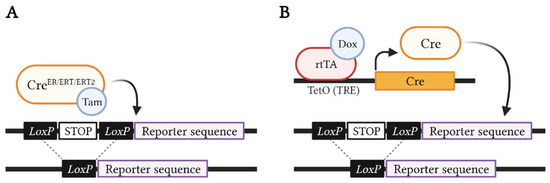
Figure 3.
Inducible Cre/Lox systems. (A) Tamoxifen-inducible Cre/Lox model, where, upon tamoxifen (Tam) administration, the cells expressing CreER, CreERT, or CreERT2 (determined by a specific promoter) will be labeled with β-galactosidase, fluorescent protein(s), or barcode sequence(s) due to the Cre-mediated recombination of the loxP sites, excising the STOP codon in the reporter transgene. (B) Tetracycline-inducible Tet/Lox system, where, upon doxycycline (Dox) administration, the cells expressing rtTA will be labeled by the chosen reporter due to the binding of the rtTA transactivator to TRE sequences (such as transcriptional activation elements (TetO)), resulting in Cre expression and consequent excision of the loxP-flanked STOP codon upstream to the reporter transgene. This figure was created with Biorender.com (accessed on 12 December 2021).
A new genetically engineered mouse model using the Flippase (Flp)/flippase recognition target (Frt) system was developed to specifically study the mammary gland [35]. The authors generated a transgenic mouse line expressing a mouse codon-optimized Flp under the control of the mouse mammary tumor virus (MMTV) promoter, which recombines Frt sequences exclusively in mammary epithelial cells. Similar to the doxycycline-dependent system, this in vivo strategy does not require the use of tamoxifen for activating Flp enzyme. Importantly, this model consists of a Cre alternative recombinase (Flp), which allows the combination of multiple site-specific recombination systems, such as Cre/loxP and Flp/Frt [35,36].
One of the most suitable approaches for lineage tracing is pulse experiments, where 16–24 h after Cre recombination, the targeted cells are analyzed; and chase experiments, which allow us to visualize the progeny of the targeted cells, genetically labeled regardless of the expression of the specific gene used for lineage tracing. Recently, some groups have used this technology to explore the capacity of different mammary epithelial cells to initiate tumors or metastasize, as well as to understand the potential mechanisms underlying cellular plasticity within breast tumors, which could ultimately lead to new therapeutic strategies for treating heterogeneous tumors in the near future.
2.1. Searching for the Tumor Cell of Origin in Different Breast Cancer Subtypes
Gene expression analyses have provided a new sub-classification of breast cancer patients based on their transcriptomic profile, which allowed them to be originally subclassified into five different molecular breast cancer subtypes: luminal A, luminal B, HER2-positive, normal-like, and basal-like [37]. Further gene expression studies identified a new breast cancer subtype known as claudin-low or mesenchymal-like [38]; and later, clustering analysis of genomic and transcriptomic data of breast tumors revealed 10 novel integrative clusters, which displayed different copy number alterations and gene expression profiles associated with distinct clinical outcomes [39].
Importantly, several studies compared the gene expression signature of different healthy mammary epithelial cells (MECs) with transcriptional profiles of different tumor subtypes [17,40,41]. Interestingly, it was found that the luminal progenitor cell signature resembled that of basal-like tumors, suggesting that the cell of origin in this breast cancer subtype could be an LC, both in mouse and human [17,40,41]. Conversely, the BC signature was upregulated in the claudin-low subtype, and the mature luminal gene signature was closely aligned with the luminal A and B subtypes [17,41]. Overall, these results introduced comparative expression profiling as a powerful tool to elucidate the cell of origin in different cancer subtypes, which could serve as a cellular target for oncogenic events; however, only genetic studies at the single-cell level or lineage tracing experiments are the current definitive approaches to identify the cell of origin in each breast cancer subtype.
So far, only a few studies have explored this particular issue using lineage tracing tools. A good example is a recent study that proved that cells positive for leucine-rich repeat-containing G-protein coupled receptor 6 (Lgr6), which is expressed in LCs and BCs during the early stages of tumorigenesis, contributed to mammary tumor progression [42]. Specifically, when Lgr6pos cells were genetically labeled at the hyperplasia stage (P12, 12-day-old) in a mouse mammary tumor virus promoter-driven Polyomavirus middle T antigen breast cancer mouse model (MMTV-PyMT), which mostly generates luminal tumors, these cells clonally expanded, contributing to the formation of carcinomas [42]. Surprisingly, when they used the Medroxyprogesterone Acetate (MPA) plus 7,12-Dimethylbenz[a]anthracene (DMBA)-dependent model to generate mixed luminal and basal tumors [42], their lineage tracing analysis showed that neither basal nor luminal Lgr6pos cells were involved in the formation of basal-like tumors. These results suggest that Lgr6pos cells could be the cell of origin in the luminal breast cancer subtype exclusively; however, the fact that the Lgr6 promoter is activated in LCs and BCs at the start point of the lineage tracing makes it complicated to draw strong conclusions regarding the true cell of origin in each breast cancer subtype. Remarkably, when Lgr5-expressing cells (exclusively expressed in BCs [43]) were traced in C3(1)Tag mice, a murine model that spontaneously develops TNBC [44,45], the hyperplastic lesions generated were mostly Lgr5pos-derived progeny, denoting Lgr5pos cells as the cellular origin of TNBCs [46]; nevertheless, an exhaustive histological characterization of the resulting tumors was missing in this work, making it difficult to conclude which specific types of tumors were formed from these cells.
Fascinatingly, a recent work combined lineage tracing analysis and the RCAS-TVA system. This consists of expressing the TVA cell surface receptor under the control of a specific target promoter recognized by an avian leukosis virus-derived vector (RCAS) [47] to elucidate the contribution of ERα during cancer progression and metastasis of HER2-positive tumors [48]. By using Esr1-Cre/MMTV-TVA/Rosa26-tdRFP mice infected with RCAS-Erbb2 (to generate HER2-positive tumors), the authors could demonstrate that ERαpos tumor cells with an overexpression of HER2 have to progressively lose their ERα expression in order to clonally expand and metastasize [48]. Importantly, HER2-positive cells originated from ERαpos cells were more aggressive than those originated from ERαneg cells, suggesting that the cell of origin plays an important role in the clinical outcome of breast cancer patients.
Undoubtedly, more studies are required to figure out which mammary epithelial cell type is the cell of origin for each specific tumor type. The lack of good preclinical breast cancer mouse models that truly recapitulate human subtypes, and the selection of suitable inducible Cre lines, which exclusively label one mammary epithelial cell type at a time, are the major concerns that the research community currently face to address this important issue.
2.2. Importance of the Epithelial-to-Mesenchymal Transition during Tumor Progression
The epithelial-to-mesenchymal transition (EMT) is a biologic process by which epithelial cells lose their cell polarity and cell–cell adhesion to undergo multiple biochemical changes that enable them to assume mesenchymal attributes, such as an elongated shape, fibroblast-like morphology, enhanced mobility, invasiveness, resistance to apoptotic stimuli, and production of extracellular matrix components [49]. Crucially, this process could be exploited by tumor cells, allowing them to detach from each other within the primary tumor and metastasize to distant organs [50].
In vivo monitorization of different mesenchymal markers (FSP1 and Vimentin) using different breast cancer models demonstrated that the EMT process does not contribute to lung metastasis development [51]. These studies evaluated the appearance of EMT in two breast cancer models (MMTV-PyMT and MMTV-Neu), being a biological process that can be observed in vivo; however, the metastases developed were neither FSP1- nor Vimentin-derived progenies [51,52,53]. Notwithstanding, FSP1-derived mesenchymal cells can undergo mesenchymal-to-epithelial transition (MET) and contribute to tumor recurrence, although this was addressed using serial transplantations as a rough way of recapitulating this tumor process [54]. The main concern regarding these studies was that these murine lines were targeting a small fraction of the total cells that undergo EMT, having a non-negligible difference between the total percentage of mesenchymal E-Cadherin (Ecad)low cells and FSP1pos-cells (5% and 0.3%, respectively) [52]. Crucially, when Ecadlow tumor cells were injected into the circulation, these cells generated metastases [52]; however, these experiments did not address the fact that this process occurs naturally in vivo during the progression of this disease.
The fact that many different cells can undergo EMT and the lack of a universal marker encouraged the combinatorial design of Cre and Dre systems to study this biological process. Recently, Li and colleagues generated EMTracer, a triple transgenic mouse model carrying Kit-CreER, EMTgene-LSL-Dre, and NR1-reporter. Thus, this EMTracer was crossed with the MMTV-PyMT model to monitor EMT during tumor progression [55]. With this strategy, after tamoxifen administration, luminal-Kitpos cells recombined LoxP sequences expressing ZsGreen fluorescent protein, inducing Dre expression exclusively in mesenchymal cells which were positive for Vim or Cdh2/N-cadherin, turning tdTomato positive [55]. In their functional assays, they found that Vimentin was not functionally required to metastasize (as previously reported); however, the activation of N-cadherin was critical for lung colonization [55]. In addition, the authors demonstrated that breast cancer cells underwent the EMT program during primary tumor growth rather than during dissemination or lung colonization [55], indicating the importance of studying EMT at different stages of the metastatic cascade to draw reliable conclusions.
The heterogeneity underlying the EMT process could be explained by the emergence of a partial rather than a full EMT [56], resulting in the appearance of intermediate hybrid states that share epithelial and mesenchymal features. In fact, combining lineage tracing strategies with single cell RNA-sequencing (scRNA-seq) analyses at different time points of the metastatic cascade could shed light on the characterization of the different EMT transitioning states that breast cancer cells undergo.
2.3. Differential Clonal Expansion during Tumor Progression and the Metastatic Process
New microscopy technologies, such as intravital or 3D-whole mount imaging, have allowed the visualization of mammary tumor progression from adenoma to carcinoma in vivo and at large-scale single-cell resolution, respectively [57,58]. Combining intravital imaging with random multi-labeling of the mammary gland (using Rosa26-CreERT2), Zomer and colleagues observed that only a small subset of tumor cells clonally expanded during tumor progression, and the vast majority of cells within the primary tumor either disappear, grow slowly, or partially expand to finally regress [57]. The combination of these technologies with cell-specific lineage tracing analysis to further study the potential of each MEC to metastasize still represents a gap in our field of knowledge.
Performing multicolor fluorescent lineage tracing in the MMTV-PyMT model demonstrated that the metastatic process is produced by the collective dissemination of cancer cells forming cohesive clusters rather than the serial seeding of single tumor cells [59]. Concretely, after the orthotopic implantation of mammary tumors formed of cells randomly labeled with different fluorescent proteins, the resulting lung metastases were formed of multicolored cells, signifying multicellular seeds [59]. Moreover, these lineage tracing studies showed multicolored tumor cell clusters at five different stages of metastasis: collective invasion, locally disseminated clusters in the adjacent stroma, extravasated tumor emboli, circulating tumor cell clusters, and distant micro- and macro-metastases [59].
A powerful approach is combining lineage tracing with scRNA-seq by introducing genetic barcodes. Indeed, using this strategy, Ginzel and colleagues measured the tumorigenic capacity of different oncogenic HER2 isoforms (HER2, d16HER2 and p95HER2) within the same mammary gland [60]. Specifically, the authors mated MMTV-Cre mice with HER2-Crainbow (HER2BOW) mice, which encoded for the three HER2 variants, fluorescently barcoded and flanked by LoxP sites. Using fluorescence and RNA sequencing, they could characterize the tumor phenotype associated with each barcoded isoform [60]. Although wild-type HER2 rarely induced indolent tumors, d16HER2 generated luminal-like proliferative in situ lesions, which eventually progressed, and p95HER2 prompted the early appearance of invasive cancers characterized by double-positive luminal and basal epithelial cells. From a clinical standpoint, these results underscore the importance of subclassifying HER2-positive breast cancer patients based on their HER2 isoform [60].
3. In Vivo Models to Study Mammary Gland Tumorigenesis
Many groups have tried to recapitulate the wide diversity of breast cancer subtypes detected in the clinics, designing preclinical mouse models that overexpress oncogenes or silence tumor suppressor genes. Indeed, recent studies have relied on Cre/lox systems for defining novel drivers of mammary tumorigenesis and assessing their consequences in different cellular contexts.
3.1. Classic Preclinical Models to Recapitulate Different Breast Cancer Subtypes
It was reported that specific murine strains can spontaneously develop breast tumors, such as CH3, that produces adenocarcinomas with a latency of 6–10 months [61]; BALB/c that also generates adenocarcinomas at 12 months of age [61]; and the Kunming strain that can develop invasive ductal carcinomas in 13.5 months [62]. In addition, there are inducible models that can be sorted by chemical treatments (DMBA or N-nitroso-N-methylurea (NMU)) [63]; physically, by the effects of radiation [64]; or biologically, by lentiviral infection [65]. However, the transcriptomic profiles of most of these models have not been fully analyzed. In order to shed light on this important issue, different research groups analyzed in depth tumors derived from different animal models by comparing their gene expression profiles with different human public databases, and calculating to what extent these models resembled the human disease [45,66]. Thus, murine models were divided into mesenchymal (also known as claudin-low subtype), basal, luminal, or HER2-enriched tumors. Among the animal models with tumors presenting mesenchymal features, mainly characterized by the expression of Vim and Snai1, they found the MMTV-Cre/Brca1Co/Co/p53+/− [67]; DMBA-induced [68], few C3(1)-Tag [44], MMTV-Lpa [69], WAP-T121 [70], and p53+/− irradiated [71] models. Recapitulating human basal-like tumors, they included MMTV-Cre/Brca1Co/Co/p53+/− [67], DMBA-induced [68], C3(1)-Tag [44], MMTV-Myc [72], WAP-Myc [73], WAP-Tag [74], MMTV-Aib1 [69], and MMTV-Wnt1 [75] mice. Resembling luminal tumors, they detected MMTV-Neu [76], MMTV-PyMT [77], WAP-Myc [73], MMTV-Myc [72], MMTV-Aib1 [69], MMTV-Hras [72], and WAP-Int3 [78]. Additionally, recapitulating HER2-enriched tumors, they found MMTV-Neu [76], Bgr1+/− [79], p18−/− [80], Rb−/− [81], MMTV-Aib1 [69], WAP-Cre/Etv6 [82], WAP-T121 [70], and MMTV-Fgf3 [83]. (Table 1).

Table 1.
Murine models that recapitulate different human breast cancer subtypes. This table summarizes the transcriptomic analysis performed in tumors derived from different murine in vivo models that are able to resemble specific human breast cancer subtypes.
Some of these models have been the in vivo approaches of reference to study breast cancer for decades; however, they are not sufficient to understand the wide spectrum of human breast tumors. Moreover, the major differences in the composition of the stroma between the murine and human mammary gland, being more adipocytic and less fibrotic in mice than in humans [84], might play an important role during tumor progression, as well as the colonization of different metastatic organs, since mouse models only develop distant lung metastases, whereas humans are able to metastasize into lung, liver, bone, or brain [85].
3.2. Cellular Plasticity Plays an Important Role in the Development of Mammary Tumors
Numerous lineage tracing studies in healthy mammary glands have demonstrated that there are no multipotent stem cells in adult mice, but distinct pools of unipotent stem cells, which self-sustain the lineage restriction of each mammary epithelial population [86,87,88]. However, adult MECs have been shown to be extremely plastic under different stress situations, such as transplantation [86], oncogene activation [18,43,89,90], cellular ablation [91], or the ectopic expression of key cell fate determinants [92,93], interconverting their cellular potency and destiny. This cellular plasticity observed in normal epithelial cells may be conceivably magnified in tumors, thus contributing to the cellular heterogeneity observed in breast cancer.
In the cancer context, two independent groups have shown that luminal ERαpos tumors can arise from BCs and LCs due to the expression of the oncogenic form PIK3CAH1047R [43,89]. In fact, PIK3CAH1047R was enough to induce cell plasticity and the acquisition of multi-lineage features, defined by the expression of both luminal and basal gene signatures at the same time [43]. Interestingly, when the same oncogenic hit was overexpressed in LCs (Krt8-expressing cells) or BCs (Lgr5-expressing cells), the resulting tumors were basal-like, HER2-positive, and luminal B (Krt8-derived tumor), or mainly luminal A and B (Lgr5-derived tumors) [43] (Figure 4). Similar results were found using the Krt5 promoter to target BCs [89]; concretely, overexpression of PIK3CAH1047R in BCs led to the formation of luminal B tumors (Figure 4), while Krt8-expressing cells generated luminal B and basal-like tumors [89]. All these results suggest that ERα-positive tumors with mutations in PI3K could originate from LCs and also BCs, whereas luminal ERα-negative, basal-like, and HER2-enriched tumors may exclusively arise from LCs. Indeed, these studies show how different the cellular plasticity is in LCs and BCs, and how the activation of a specific oncogene can give rise to different latency and tumor types depending on the cell of origin.
In the same direction, the loss of Brca1 and Tp53 in different cellular compartments resulted in the development of different breast cancer subtypes [18]. In this particular work, the authors used Krt14-Cre or Blg-Cre lines to monitor BCs or ERαneg LCs, respectively, and mated them with Brca1fl/fl/p53+/− mice. Importantly, BRCA1/p53 deficiency generated different types of tumors that histologically expressed basal markers (Keratin-14 or p63) and luminal markers (Keratin-18 and/or ERα), with similar but not identical gene expression profiles, which closely resembled that of human basal-like tumors, regardless of the cell of origin (Figure 4). Importantly, only Bgl-derived tumors histologically resembled human BRCA1 loss-of-function [18].
Strikingly, using the luminal Cre line Wap-Cre, different groups were able to observe a luminal-to-basal conversion, either overexpressing NTRK3 [94], active NOTCH1 (N1ICD) [95], or KRASG12D [96]. NTRK3 was able to induce mixed tumors bearing both basal and luminal cells, as well as hybrid tumors with cells expressing basal and luminal markers simultaneously [94]; N1ICD specifically generated tumors that transcriptionally matched with distinct luminal subtypes, but also with a mixed subtype containing luminal and basal identities [95], and the exogenous expression of mutant KRASG12D led to metastatic claudin-low mammary tumors with a mesenchymal-like phenotype [96] (Figure 4).

Figure 4.
Schematic representation of the mammary cellular plasticity during tumorigenesis. The use of different murine Cre lines (Lgr5-CreERT2, Krt5-CreERT2 or Krt14-Cre to target BCs; Bgl-Cre or Wap-Cre to trace ERαneg LCs; and Esr1-Cre to monitor ERαpos LCs) to induce the expression of specific mutations in different mammary epithelial cell types resulted in the generation of tumors that resembled some of the human subtypes: luminal A [43], luminal B [43,89], HER2-enriched [48,66], claudin-low (or mesenchymal-like) [96], and basal-like [18,73] tumors. Notes: this figure only includes studies that have used comparisons with human breast cancer transcriptomic datasets (PAM50, Hu306 or similar); studies performed with non-specific Cre lines, such as MMTV-Cre or Krt8-CreERT2 (which labels all MECs, or both LC populations, respectively), were discarded. This figure was created with Biorender.com (accessed on 12 December 2021).
Another interesting strategy to study cellular plasticity is the use of the RCAS-TVA system [47]. Using this retrovirus-mediated in vivo lineage tracing, Hein and colleagues engineered RCAS vectors bearing either PyMT or HER2 constructs, which were produced and infected in Wap-TVA transgenic mice that specifically express the TVA receptor in LCs [90]. Similar to the aforementioned studies, the activation of certain oncogenes in the luminal compartment was sufficient to develop tumors with either luminal, basal, or mixed (Keratin-8 and Keratin-5-positive) features [90].
Collectively, these studies have demonstrated that cellular plasticity could be responsible for the intra- and inter-tumor heterogeneity found in breast cancer and pose new questions such as whether any mammary epithelial cell has the potential to become a tumor-initiating cell, or, by contrast, only a suitable combination of oncogenic hits is the crucial determinant in developing a tumor regardless of the cell of origin. Hence, these findings emphasize the importance of searching for key factors underlying cellular plasticity, which could have significant implications for cancer therapeutics.
4. Clinical Implications of Lineage Tracing Studies and Future Perspectives
Clinical decisions are made depending on the molecular subtype diagnosed, which includes anti-hormone therapies for those patients with ERαpos cells, anti-HER2 treatments for HER2-positive patients, and chemotherapy for patients diagnosed with TNBC, due to the lack of specific targets [2]. These therapeutic strategies do not take into account that tumors are heterogeneous, meaning that they are composed of different types of cells, which could be the main cause of clinical failures. In this sense, the main handicap that researchers are currently facing is the lack of reliable preclinical models that resemble this human intratumor heterogeneity. Currently, the vast majority of in vivo mammary tumorigenesis models available depend on the expression of a specific driver, which eventually contributes to the formation of practically (intra)homogeneous tumors. The combination of multiple in vivo systems (Cre/lox, RCAS/TVA, Dre/rox, and Flp/Frt) could be the key to designing a model able to recapitulate the tumor evolution observed in human breast cancer.
Remarkably, lineage tracing studies have disclosed that the cell of origin matters. Here, we have presented several good examples, such as the work of Ding and colleagues that proved that HER2-positive tumors with an ERαpos or ERαneg cellular origin will determine their aggressiveness and metastatic capacity [48]. Moreover, these in vivo approaches have also revealed an inherent cellular plasticity in MECs; along this line, different groups have demonstrated that upon the activation of a specific oncogenic hit, MECs can acquire new transcriptomic features and expand to develop tumors with different cell fate signatures [18,43,89,94,95,96]. Some of these studies tried to understand whether the cell of origin or the oncogenic activation were playing a major role during tumorigenesis, and concluded that the cell of origin was crucial for determining the tumor subtype and/or aggressiveness; however, in all cases, those specific oncogenes (PI3KCA [43,89] or HER2 [48]) were able to generate tumors, suggesting that both facts (cell of origin and oncogenic hit) are equally important. Numerous studies on the normal mammary gland have demonstrated the high plasticity of BCs upon different stressors (transplantation or cell ablation [86,91]) compared with LCs; nevertheless, in the cancer context, all these studies demonstrated that LCs are more plastic than BCs, being able to generate a wider range of breast tumor subtypes [18,43,89,94,95,96], representing a new field to be explored.
Human cells contain somatic mutations that have served as genetic barcodes to perform retrospective lineage tracing analysis in healthy and diseased human tissues [97]. For example, topographic single-cell sequencing from laser-capture microdissected breast cancer samples at different tumorigenic stages, analysis of copy number alterations and clonal dynamics of different areas suggested a multiclonal invasion model for breast cancer [98]. Another strategy is the use of mitochondrial DNA mutations as a natural barcode, which McDonald and colleagues used to find common mutations that would indicate a common cell of origin in both normal and premalignant breast sections [99]. Alternatively, the use of DNA barcodes, introduced by infection in isolated normal human mammary cells, revealed a complex clonal landscape within heterogeneous breast tumors expressing KRASG12D [100].
Notably, over the last years, single-cell RNA sequencing has emerged as a new tool to replace lineage tracing studies, since it provides a recapitulation of the clonal dynamics of different cell populations within a tumor in a retrospective manner; however, the question of the identity of the tumor-initiating cell still remains to be solved with this type of technology. Combining both approaches would allow us to genetically and phenotypically trace each individual cell, redefining the phylogenetic trees, cell trajectories, and cell–cell interactions. In keeping with recent breakthroughs, the development of barcode systems has enabled us to target individual cells with unique nucleic acid sequences [101]. This technology could be used for lineage tracing together with the sc-RNAseq technique to reveal the transcriptomics of each cell population within heterogeneous mammary tumors, and also to perform high-throughput genetic screening to discover key plasticity factors and tumor drivers, being potential druggable targets for breast cancer therapeutics [102]. For example, Ying and Beronja employed long-term lineage tracing using stable barcodes to study mammary tissue hierarchy, ensuring each progenitor was labeled with a single barcode [102], and designed a large-scale genetic screening with a barcoded lentivirus library that targeted multiple clinically relevant mutations [102]. Definitively, the combination of multiple technologies will allow us to identify new crucial biomarkers and novel therapeutic targets, especially for TNBC patients who have no targeted therapies available.
Beyond lineage tracing, multi-omic and high-throughput methods will help us to understand the complex genomics that underlie the human cancer disease. Indeed, single-cell genomics has enabled us to associate specific genetic mutations with different molecular subtypes [103], and single-cell transcriptomics has provided cancer-specific gene signatures that could help clinicians to determine the prognosis of patients [104]. For instance, there are currently different gene expression profiling tests for breast cancer in clinics, such as MammaPrint® and Oncotype DX®, both of which predict the risk of distant disease recurrence. Moreover, among some spatial transcriptomic technologies, fluorescence in situ hybridization (FISH) methods are commonly used in cancer diagnosis as they allow the detection and chromosomal location of specific genes which are aberrantly expressed or harbor rare mutations in tumors [105]. On the other hand, there are other techniques such as laser capture microdissection and photoactivatable transcriptome in vivo analysis that could be combined with sequencing-based approaches to decipher the genetic information of the desired area of the tissue [105]. The integration of all these spatial data promises the identification of multiple reliable molecular biomarkers that would be helpful for the diagnostics and therapeutics of breast cancer patients in the near future.
Author Contributions
E.V.-P. and D.O.-Á. performed the literature search and drafted the work. V.R. had the idea for the article and critically revised the work. All authors have read and agreed to the published version of the manuscript.
Funding
The research leading to this work received funding from the Spanish Ministry of Science abd Innovation (MCI), PID2020-117212RB-I00/AEI/10.13039/501100011033 and RYC2018-024099-I. We thank the CERCA Program of the Generalitat de Catalunya for its institutional public support, and the Departament de Salut for funding E.V.-P. with a PERIS PIF-Salut fellowship (SLT017/20/000140).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Fu, N.Y.; Nolan, E.; Lindeman, G.J.; Visvader, J.E. Stem Cells and the Differentiation Hierarchy in Mammary Gland Development. Physiol. Rev. 2020, 100, 489–523. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Li, J.; Wang, C.; Lou, Z.; Gao, S.; Zhao, L.; Wang, S.; Chaulagain, A.; Zhang, M.; Li, X.; et al. Single cell RNA sequencing for breast cancer: Present and future. Cell Death Discov. 2021, 7, 104. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, A.; Kuperwasser, C. The origin of breast tumor heterogeneity. Oncogene 2015, 34, 5309–5316. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Vassilopoulos, A.; Chisholm, C.; Lahusen, T.; Zheng, H.; Deng, C.X. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene 2014, 33, 5477–5482. [Google Scholar] [CrossRef]
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef]
- Yang, L.; Tang, H.; Kong, Y.; Xie, X.; Chen, J.; Song, C.; Liu, X.; Ye, F.; Li, N.; Wang, N.; et al. LGR5 Promotes Breast Cancer Progression and Maintains Stem-Like Cells Through Activation of Wnt/beta-Catenin Signaling. Stem Cells 2015, 33, 2913–2924. [Google Scholar] [CrossRef]
- Hwang-Verslues, W.W.; Kuo, W.H.; Chang, P.H.; Pan, C.C.; Wang, H.H.; Tsai, S.T.; Jeng, Y.M.; Shew, J.Y.; Kung, J.T.; Chen, C.H.; et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS ONE 2009, 4, e8377. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Vaillant, F.; Asselin-Labat, M.L.; Shackleton, M.; Forrest, N.C.; Lindeman, G.J.; Visvader, J.E. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008, 68, 7711–7717. [Google Scholar] [CrossRef]
- Tiede, B.; Kang, Y. From milk to malignancy: The role of mammary stem cells in development, pregnancy and breast cancer. Cell Res. 2011, 21, 245–257. [Google Scholar] [CrossRef]
- Barriga, F.M.; Montagni, E.; Mana, M.; Mendez-Lago, M.; Hernando-Momblona, X.; Sevillano, M.; Guillaumet-Adkins, A.; Rodriguez-Esteban, G.; Buczacki, S.J.A.; Gut, M.; et al. Mex3a Marks a Slowly Dividing Subpopulation of Lgr5+ Intestinal Stem Cells. Cell Stem Cell 2017, 20, 801–816.e7. [Google Scholar] [CrossRef] [PubMed]
- Tetteh, P.W.; Basak, O.; Farin, H.F.; Wiebrands, K.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; de Sauvage, F.; van Es, J.H.; et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell 2016, 18, 203–213. [Google Scholar] [CrossRef]
- Yu, S.; Tong, K.; Zhao, Y.; Balasubramanian, I.; Yap, G.S.; Ferraris, R.P.; Bonder, E.M.; Verzi, M.P.; Gao, N. Paneth Cell Multipotency Induced by Notch Activation following Injury. Cell Stem Cell 2018, 23, 46–59.e5. [Google Scholar] [CrossRef] [PubMed]
- Ayyaz, A.; Kumar, S.; Sangiorgi, B.; Ghoshal, B.; Gosio, J.; Ouladan, S.; Fink, M.; Barutcu, S.; Trcka, D.; Shen, J.; et al. Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 2019, 569, 121–125. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef]
- Danielian, P.S.; White, R.; Hoare, S.A.; Fawell, S.E.; Parker, M.G. Identification of residues in the estrogen receptor that confer differential sensitivity to estrogen and hydroxytamoxifen. Mol. Endocrinol. 1993, 7, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Brocard, J.; Mascrez, B.; LeMeur, M.; Metzger, D.; Chambon, P. Ligand-activated site-specific recombination in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10887–10890. [Google Scholar] [CrossRef]
- Metzger, D.; Clifford, J.; Chiba, H.; Chambon, P. Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc. Natl. Acad. Sci. USA 1995, 92, 6991–6995. [Google Scholar] [CrossRef]
- Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 1999, 21, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Fujiwara, Y.; Chapdelaine, A.; Yang, H.; Orkin, S.H. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood 2001, 97, 324–326. [Google Scholar] [CrossRef]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Snippert, H.J.; van der Flier, L.G.; Sato, T.; van Es, J.H.; van den Born, M.; Kroon-Veenboer, C.; Barker, N.; Klein, A.M.; van Rheenen, J.; Simons, B.D.; et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010, 143, 134–144. [Google Scholar] [CrossRef]
- Peikon, I.D.; Gizatullina, D.I.; Zador, A.M. In vivo generation of DNA sequence diversity for cellular barcoding. Nucleic Acids Res. 2014, 42, e127. [Google Scholar] [CrossRef]
- Pei, W.; Feyerabend, T.B.; Rossler, J.; Wang, X.; Postrach, D.; Busch, K.; Rode, I.; Klapproth, K.; Dietlein, N.; Quedenau, C.; et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548, 456–460. [Google Scholar] [CrossRef]
- Shehata, M.; van Amerongen, R.; Zeeman, A.L.; Giraddi, R.R.; Stingl, J. The influence of tamoxifen on normal mouse mammary gland homeostasis. Breast Cancer Res. 2014, 16, 411. [Google Scholar] [CrossRef]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed]
- Wuidart, A.; Ousset, M.; Rulands, S.; Simons, B.D.; Van Keymeulen, A.; Blanpain, C. Quantitative lineage tracing strategies to resolve multipotency in tissue-specific stem cells. Genes Dev. 2016, 30, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Luond, F.; Bill, R.; Vettiger, A.; Oller, H.; Pelczar, P.; Christofori, G. A Transgenic MMTV-Flippase Mouse Line for Molecular Engineering in Mammary Gland and Breast Cancer Mouse Models. J. Mammary Gland. Biol. Neoplasia 2019, 24, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Radler, P.D.; Vistisen, K.; Triplett, A.A.; Dennaoui, R.; Li, Y.; Shrestha, H.; Ferraiuolo, R.M.; Thangasamy, A.; Saur, D.; Wagner, K.U. Dual recombinase action in the normal and neoplastic mammary gland epithelium. Sci. Rep. 2021, 11, 20775. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef]
- Dawson, S.J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013, 32, 617–628. [Google Scholar] [CrossRef]
- Shehata, M.; Teschendorff, A.; Sharp, G.; Novcic, N.; Russell, I.A.; Avril, S.; Prater, M.; Eirew, P.; Caldas, C.; Watson, C.J.; et al. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 2012, 14, R134. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Pervolarakis, N.; Blake, K.; Ma, D.; Davis, R.T.; James, N.; Phung, A.T.; Willey, E.; Kumar, R.; Jabart, E.; et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018, 9, 2028. [Google Scholar] [CrossRef] [PubMed]
- Blaas, L.; Pucci, F.; Messal, H.A.; Andersson, A.B.; Josue Ruiz, E.; Gerling, M.; Douagi, I.; Spencer-Dene, B.; Musch, A.; Mitter, R.; et al. Lgr6 labels a rare population of mammary gland progenitor cells that are able to originate luminal mammary tumours. Nat. Cell Biol. 2016, 18, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O.; et al. PIK3CA(H1047R) induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef]
- Maroulakou, I.G.; Anver, M.; Garrett, L.; Green, J.E. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc. Natl. Acad. Sci. USA 1994, 91, 11236–11240. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef]
- Hagerling, C.; Owyong, M.; Sitarama, V.; Wang, C.Y.; Lin, C.; van den Bijgaart, R.J.E.; Koopman, C.D.; Brenot, A.; Nanjaraj, A.; Warnberg, F.; et al. LGR5 in breast cancer and ductal carcinoma in situ: A diagnostic and prognostic biomarker and a therapeutic target. BMC Cancer 2020, 20, 542. [Google Scholar] [CrossRef]
- Du, Z.; Podsypanina, K.; Huang, S.; McGrath, A.; Toneff, M.J.; Bogoslovskaia, E.; Zhang, X.; Moraes, R.C.; Fluck, M.; Allred, D.C.; et al. Introduction of oncogenes into mammary glands in vivo with an avian retroviral vector initiates and promotes carcinogenesis in mouse models. Proc. Natl. Acad. Sci. USA 2006, 103, 17396–17401. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, Y.; Lee, D.K.; Tong, Z.; Yu, X.; Li, Y.; Xu, Y.; Lanz, R.B.; O’Malley, B.W.; Xu, J. Cell lineage tracing links ERalpha loss in Erbb2-positive breast cancers to the arising of a highly aggressive breast cancer subtype. Proc. Natl. Acad. Sci. USA 2021, 118, e2100673118. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Bornes, L.; van Scheppingen, R.H.; Beerling, E.; Schelfhorst, T.; Ellenbroek, S.I.J.; Seinstra, D.; van Rheenen, J. Fsp1-Mediated Lineage Tracing Fails to Detect the Majority of Disseminating Cells Undergoing EMT. Cell Rep. 2019, 29, 2565–2569.e2563. [Google Scholar] [CrossRef]
- Sikandar, S.S.; Kuo, A.H.; Kalisky, T.; Cai, S.; Zabala, M.; Hsieh, R.W.; Lobo, N.A.; Scheeren, F.A.; Sim, S.; Qian, D.; et al. Role of epithelial to mesenchymal transition associated genes in mammary gland regeneration and breast tumorigenesis. Nat. Commun. 2017, 8, 1669. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, A.R.; Ban, Y.; Crowley, M.J.; Lee, S.B.; Ramchandani, D.; Du, W.; Elemento, O.; George, J.T.; Jolly, M.K.; Levine, H.; et al. Differential Contributions of Pre- and Post-EMT Tumor Cells in Breast Cancer Metastasis. Cancer Res. 2020, 80, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lv, Z.; Zhang, S.; Wang, Z.; He, L.; Tang, M.; Pu, W.; Zhao, H.; Zhang, Z.; Shi, Q.; et al. Genetic Fate Mapping of Transient Cell Fate Reveals N-Cadherin Activity and Function in Tumor Metastasis. Dev. Cell 2020, 54, 593–607.e5. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Zomer, A.; Ellenbroek, S.I.; Ritsma, L.; Beerling, E.; Vrisekoop, N.; Van Rheenen, J. Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem Cells 2013, 31, 602–606. [Google Scholar] [CrossRef]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; Group, D.; et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019, 35, 618–632.e6. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef]
- Ginzel, J.D.; Acharya, C.R.; Lubkov, V.; Mori, H.; Boone, P.G.; Rochelle, L.K.; Roberts, W.L.; Everitt, J.I.; Hartman, Z.C.; Crosby, E.J.; et al. HER2 Isoforms Uniquely Program Intratumor Heterogeneity and Predetermine Breast Cancer Trajectories During the Occult Tumorigenic Phase. Mol. Cancer Res. 2021, 19, 1699–1711. [Google Scholar] [CrossRef]
- Heston, W.E.; Vlahakis, G. Mammary tumors, plaques, and hyperplastic alveolar nodules in various combinations of mouse inbred strains and the different lines of the mammary tumor virus. Int. J. Cancer 1971, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhou, B.; Meng, X.; Zhu, W.; Zuo, A.; Wang, X.; Jiang, R.; Yu, S. A model of spontaneous mouse mammary tumor for human estrogen receptor- and progesterone receptor-negative breast cancer. Int. J. Oncol. 2014, 45, 2241–2249. [Google Scholar] [CrossRef]
- Chan, M.M.; Lu, X.; Merchant, F.M.; Iglehart, J.D.; Miron, P.L. Serial transplantation of NMU-induced rat mammary tumors: A model of human breast cancer progression. Int. J. Cancer 2007, 121, 474–485. [Google Scholar] [CrossRef]
- Russo, I.H.; Russo, J. Mammary gland neoplasia in long-term rodent studies. Environ. Health Perspect. 1996, 104, 938–967. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.H.; Orsulic, S.; Holland, E.; Hively, W.P.; Li, Y.; Lewis, B.C.; Williams, B.O.; Varmus, H.E. Development of a flexible and specific gene delivery system for production of murine tumor models. Oncogene 1999, 18, 5253–5260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pfefferle, A.D.; Herschkowitz, J.I.; Usary, J.; Harrell, J.C.; Spike, B.T.; Adams, J.R.; Torres-Arzayus, M.I.; Brown, M.; Egan, S.E.; Wahl, G.M.; et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 2013, 14, R125. [Google Scholar] [CrossRef]
- Xu, X.; Wagner, K.U.; Larson, D.; Weaver, Z.; Li, C.; Ried, T.; Hennighausen, L.; Wynshaw-Boris, A.; Deng, C.X. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat. Genet. 1999, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Bai, R.; Russell, R.G.; Beildeck, M.E.; Xie, Z.; Kopelovich, L.; Glazer, R.I. Characterization of medroxyprogesterone and DMBA-induced multilineage mammary tumors by gene expression profiling. Mol. Carcinog. 2005, 44, 42–50. [Google Scholar] [CrossRef]
- Torres-Arzayus, M.I.; Font de Mora, J.; Yuan, J.; Vazquez, F.; Bronson, R.; Rue, M.; Sellers, W.R.; Brown, M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell 2004, 6, 263–274. [Google Scholar] [CrossRef]
- Simin, K.; Wu, H.; Lu, L.; Pinkel, D.; Albertson, D.; Cardiff, R.D.; Van Dyke, T. pRb inactivation in mammary cells reveals common mechanisms for tumor initiation and progression in divergent epithelia. PLoS Biol. 2004, 2, E22. [Google Scholar] [CrossRef]
- Backlund, M.G.; Trasti, S.L.; Backlund, D.C.; Cressman, V.L.; Godfrey, V.; Koller, B.H. Impact of ionizing radiation and genetic background on mammary tumorigenesis in p53-deficient mice. Cancer Res. 2001, 61, 6577–6582. [Google Scholar]
- Sinn, E.; Muller, W.; Pattengale, P.; Tepler, I.; Wallace, R.; Leder, P. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: Synergistic action of oncogenes in vivo. Cell 1987, 49, 465–475. [Google Scholar] [CrossRef]
- Sandgren, E.P.; Schroeder, J.A.; Qui, T.H.; Palmiter, R.D.; Brinster, R.L.; Lee, D.C. Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res. 1995, 55, 3915–3927. [Google Scholar] [PubMed]
- Husler, M.R.; Kotopoulis, K.A.; Sundberg, J.P.; Tennent, B.J.; Kunig, S.V.; Knowles, B.B. Lactation-induced WAP-SV40 Tag transgene expression in C57BL/6J mice leads to mammary carcinoma. Transgenic Res. 1998, 7, 253–263. [Google Scholar] [CrossRef]
- Tsukamoto, A.S.; Grosschedl, R.; Guzman, R.C.; Parslow, T.; Varmus, H.E. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell 1988, 55, 619–625. [Google Scholar] [CrossRef]
- Guy, C.T.; Webster, M.A.; Schaller, M.; Parsons, T.J.; Cardiff, R.D.; Muller, W.J. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10578–10582. [Google Scholar] [CrossRef]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Gallahan, D.; Jhappan, C.; Robinson, G.; Hennighausen, L.; Sharp, R.; Kordon, E.; Callahan, R.; Merlino, G.; Smith, G.H. Expression of a truncated Int3 gene in developing secretory mammary epithelium specifically retards lobular differentiation resulting in tumorigenesis. Cancer Res. 1996, 56, 1775–1785. [Google Scholar] [PubMed]
- Bultman, S.J.; Herschkowitz, J.I.; Godfrey, V.; Gebuhr, T.C.; Yaniv, M.; Perou, C.M.; Magnuson, T. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene 2008, 27, 460–468. [Google Scholar] [CrossRef]
- Pei, X.H.; Bai, F.; Smith, M.D.; Usary, J.; Fan, C.; Pai, S.Y.; Ho, I.C.; Perou, C.M.; Xiong, Y. CDK inhibitor p18(INK4c) is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell 2009, 15, 389–401. [Google Scholar] [CrossRef]
- Jiang, Z.; Deng, T.; Jones, R.; Li, H.; Herschkowitz, J.I.; Liu, J.C.; Weigman, V.J.; Tsao, M.S.; Lane, T.F.; Perou, C.M.; et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Investig. 2010, 120, 3296–3309. [Google Scholar] [CrossRef]
- Li, Z.; Tognon, C.E.; Godinho, F.J.; Yasaitis, L.; Hock, H.; Herschkowitz, J.I.; Lannon, C.L.; Cho, E.; Kim, S.J.; Bronson, R.T.; et al. ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer Cell 2007, 12, 542–558. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.J.; Lee, F.S.; Dickson, C.; Peters, G.; Pattengale, P.; Leder, P. The int-2 gene product acts as an epithelial growth factor in transgenic mice. EMBO J. 1990, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Cardiff, R.D.; Wellings, S.R. The comparative pathology of human and mouse mammary glands. J. Mammary Gland. Biol. Neoplasia 1999, 4, 105–122. [Google Scholar] [CrossRef]
- Giles, E.D.; Wellberg, E.A. Preclinical Models to Study Obesity and Breast Cancer in Females: Considerations, Caveats, and Tools. J. Mammary Gland. Biol. Neoplasia 2020, 25, 237–253. [Google Scholar] [CrossRef]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Rodilla, V.; Dasti, A.; Huyghe, M.; Lafkas, D.; Laurent, C.; Reyal, F.; Fre, S. Luminal progenitors restrict their lineage potential during mammary gland development. PLoS Biol. 2015, 13, e1002069. [Google Scholar] [CrossRef]
- Wang, C.; Christin, J.R.; Oktay, M.H.; Guo, W. Lineage-Biased Stem Cells Maintain Estrogen-Receptor-Positive and -Negative Mouse Mammary Luminal Lineages. Cell Rep. 2017, 18, 2825–2835. [Google Scholar] [CrossRef]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohee, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef]
- Hein, S.M.; Haricharan, S.; Johnston, A.N.; Toneff, M.J.; Reddy, J.P.; Dong, J.; Bu, W.; Li, Y. Luminal epithelial cells within the mammary gland can produce basal cells upon oncogenic stress. Oncogene 2016, 35, 1461–1467. [Google Scholar] [CrossRef]
- Centonze, A.; Lin, S.; Tika, E.; Sifrim, A.; Fioramonti, M.; Malfait, M.; Song, Y.; Wuidart, A.; Van Herck, J.; Dannau, A.; et al. Heterotypic cell-cell communication regulates glandular stem cell multipotency. Nature 2020, 584, 608–613. [Google Scholar] [CrossRef]
- Lilja, A.M.; Rodilla, V.; Huyghe, M.; Hannezo, E.; Landragin, C.; Renaud, O.; Leroy, O.; Rulands, S.; Simons, B.D.; Fre, S. Clonal analysis of Notch1-expressing cells reveals the existence of unipotent stem cells that retain long-term plasticity in the embryonic mammary gland. Nat. Cell Biol. 2018, 20, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Wuidart, A.; Sifrim, A.; Fioramonti, M.; Matsumura, S.; Brisebarre, A.; Brown, D.; Centonze, A.; Dannau, A.; Dubois, C.; Van Keymeulen, A.; et al. Early lineage segregation of multipotent embryonic mammary gland progenitors. Nat. Cell Biol. 2018, 20, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; van Bragt, M.P.; Li, Z. A Long-Lived Luminal Subpopulation Enriched with Alveolar Progenitors Serves as Cellular Origin of Heterogeneous Mammary Tumors. Stem Cell Rep. 2015, 5, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Phoon, Y.P.; Chivukula, I.V.; Tsoi, Y.L.; Kanatani, S.; Uhlen, P.; Kuiper, R.; Lendahl, U. Notch activation in the mouse mammary luminal lineage leads to ductal hyperplasia and altered partitioning of luminal cell subtypes. Exp. Cell Res. 2020, 395, 112156. [Google Scholar] [CrossRef]
- Radler, P.D.; Wehde, B.L.; Triplett, A.A.; Shrestha, H.; Shepherd, J.H.; Pfefferle, A.D.; Rui, H.; Cardiff, R.D.; Perou, C.M.; Wagner, K.U. Highly metastatic claudin-low mammary cancers can originate from luminal epithelial cells. Nat. Commun. 2021, 12, 3742. [Google Scholar] [CrossRef]
- Baron, C.S.; van Oudenaarden, A. Unravelling cellular relationships during development and regeneration using genetic lineage tracing. Nat. Rev. Mol. Cell Biol. 2019, 20, 753–765. [Google Scholar] [CrossRef]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217.e12. [Google Scholar] [CrossRef]
- Cereser, B.; Jansen, M.; Austin, E.; Elia, G.; McFarlane, T.; van Deurzen, C.H.; Sieuwerts, A.M.; Daidone, M.G.; Tadrous, P.J.; Wright, N.A.; et al. Analysis of clonal expansions through the normal and premalignant human breast epithelium reveals the presence of luminal stem cells. J. Pathol. 2018, 244, 61–70. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Pellacani, D.; Lefort, S.; Kannan, N.; Osako, T.; Makarem, M.; Cox, C.L.; Kennedy, W.; Beer, P.; Carles, A.; et al. Barcoding reveals complex clonal dynamics of de novo transformed human mammary cells. Nature 2015, 528, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Kebschull, J.M.; Zador, A.M. Cellular barcoding: Lineage tracing, screening and beyond. Nat. Methods 2018, 15, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Beronja, S. Embryonic Barcoding of Equipotent Mammary Progenitors Functionally Identifies Breast Cancer Drivers. Cell Stem Cell 2020, 26, 403–419.e4. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.; Purohit, P.; Misra, R.; Lingeswaran, M.; Vishnoi, J.R.; Pareek, P.; Misra, S.; Sharma, P. Single Cell Omics of Breast Cancer: An Update on Characterization and Diagnosis. Indian J. Clin. Biochem. 2019, 34, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Asselin-Labat, M.L.; Nguyen, Q.; Berthelet, J.; Tan, X.; Wimmer, V.C.; Merino, D.; Rogers, K.L.; Naik, S.H. Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods 2021, 18, 997–1012. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).