
Unveiling Metabolic Vulnerability and Plasticity of Human Osteosarcoma Stem and Differentiated Cells to Improve Cancer Therapy

 , , ,
, , ,  , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Metabolic Flux Analysis
2.3. Mitochondrial DNA Quantification
2.4. Live Cell Imaging of mtΔΨ, ROS, and Mitochondrial Morphology
2.5. Real-Time Monitoring of Cell Growth by the xCELLigence System
2.6. Apoptosis Assay and Cell-Cycle Analysis
2.7. Western Blotting Analysis
2.8. Statistical Analysis
3. Results
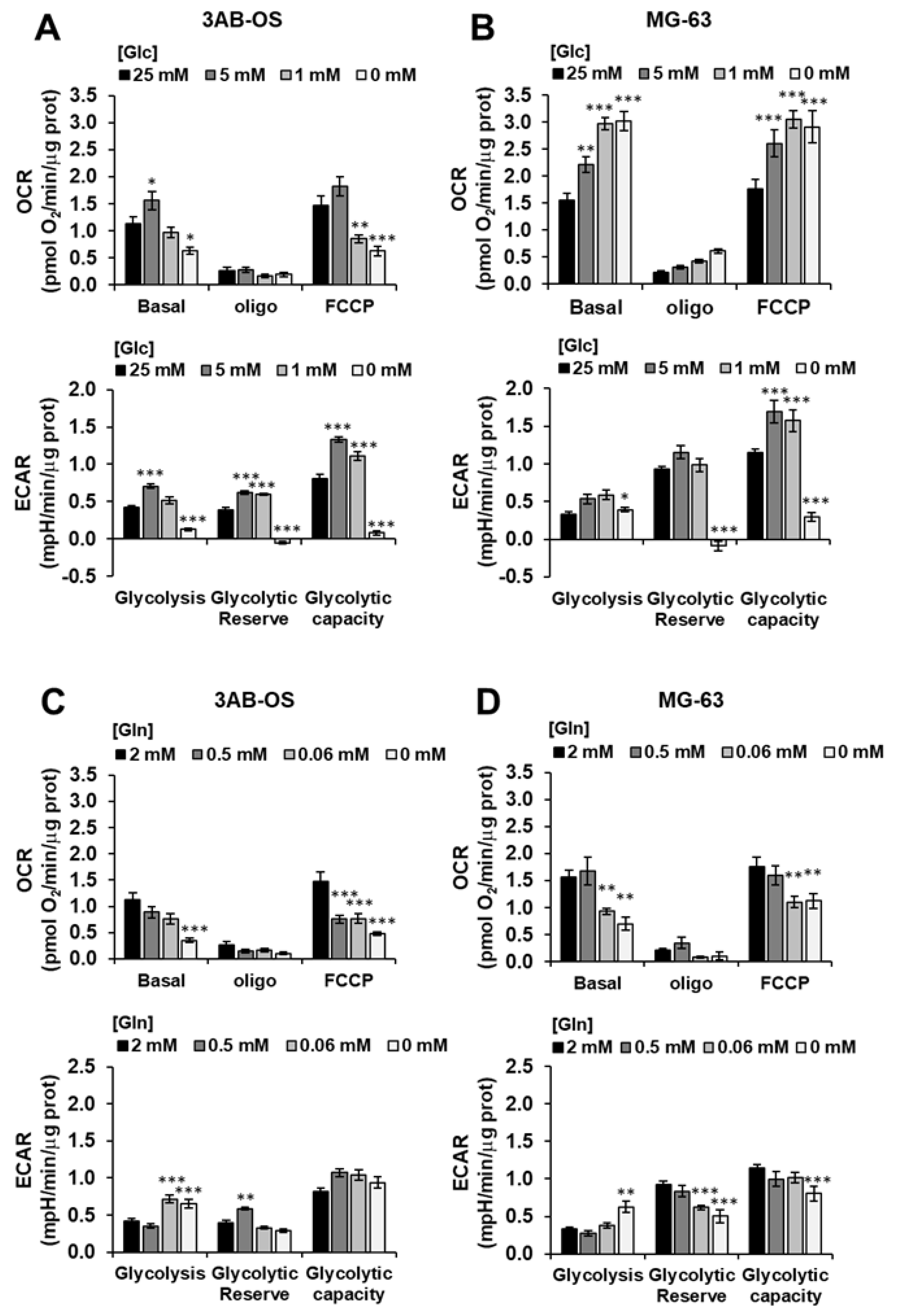
3.1. Metabolic Profiling of 3AB-OS Cancer Stem Cells and MG-63 Differentiated Cancer Cells
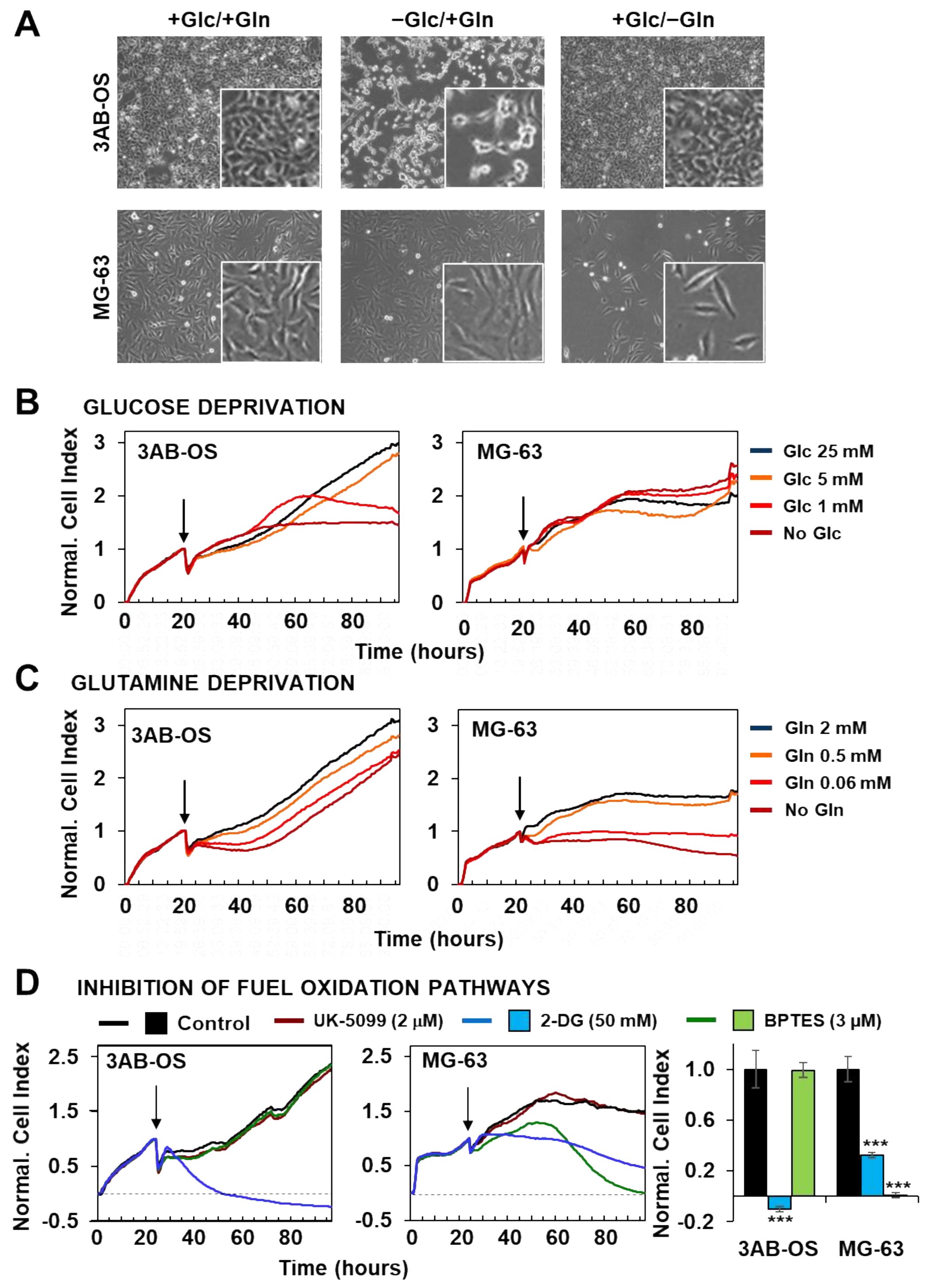
3.2. Effects of Energy Substrate Availability on Tumor Cell Proliferation
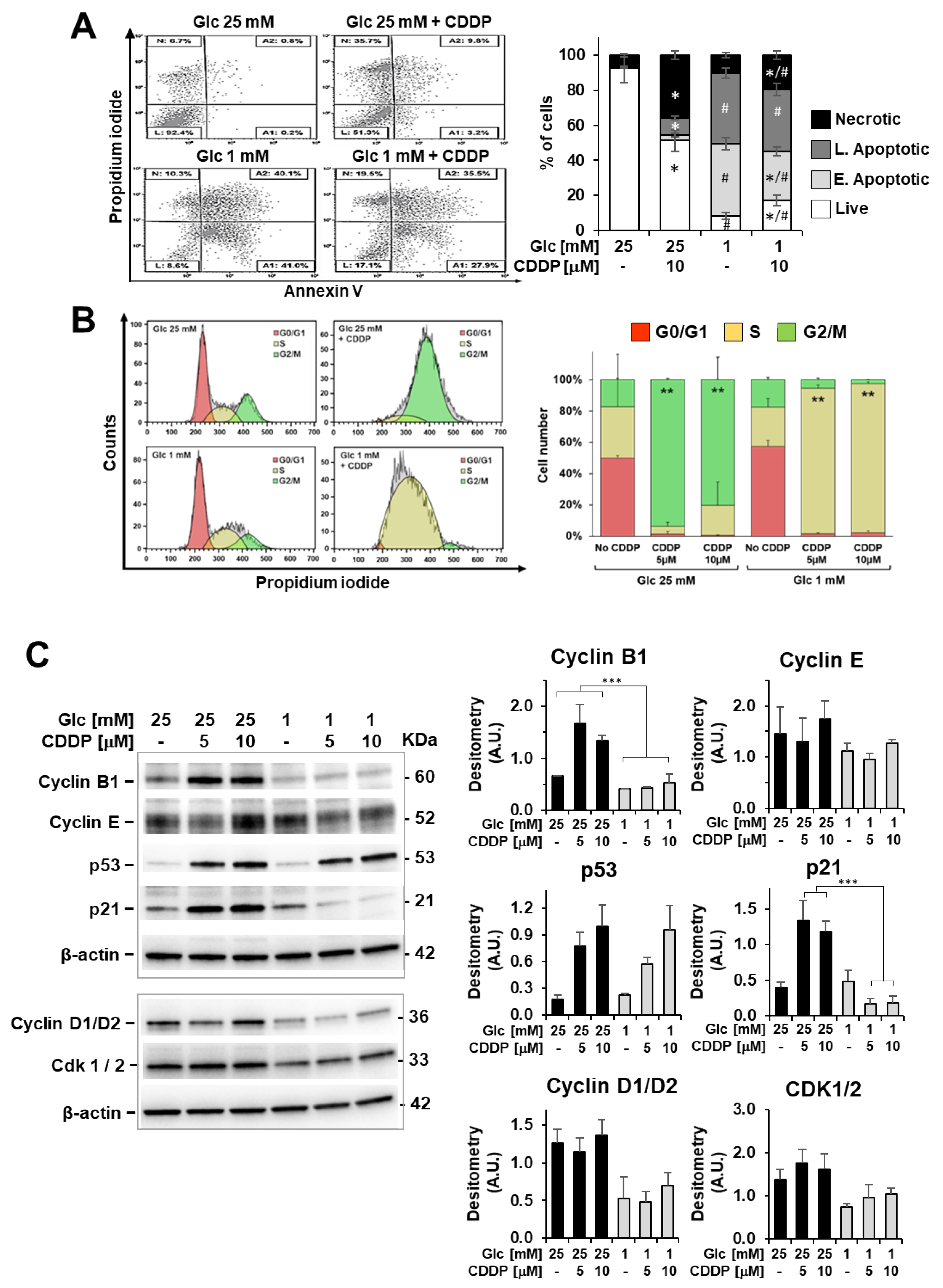
3.3. Cell Viability Effects of Cisplatin Combined with Energy Substrate Deprivation
3.4. Cisplatin Attenuates Sensitivity to Low Glucose as Well as Glucose Shortage Elicits Cisplatin Resistance
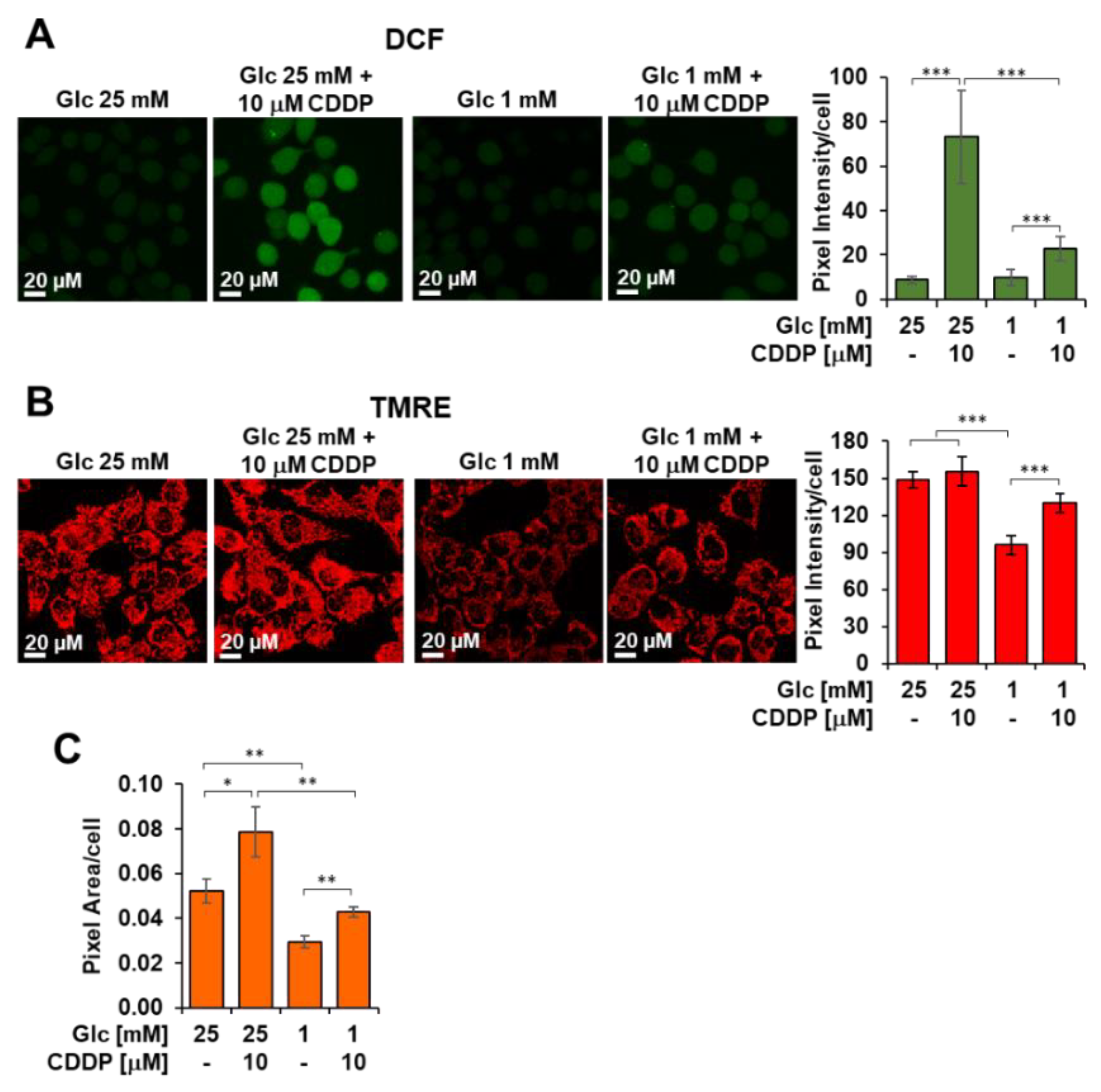
3.5. Cisplatin Enhances Glycolysis, Mitochondrial OxPhos and Metabolic Flexibility in 3AB-OS CSCs
3.6. Glucose Deprivation Induces Activation of Autophagy to Sustain Cell Proliferation
4. Discussion
4.1. Metabolic Heterogeneity of 3AB-OS and MG63 Osteosarcoma Cells Requires Distinct Therapeutic Approaches
4.2. Cisplatin Enhances 3AB-OS CSCs Metabolic Plasticity and Resistance to Glucose Starvation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, C.; Peng, K.; Guo, H.; Ren, X.; Hu, S.; Cai, Y.; Han, Y.; Ma, L.; Xu, P. miR-18a-5p promotes cell invasion and migration of osteosarcoma by directly targeting IRF2. Oncol. Lett. 2018, 16, 3150–3156. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, S.; Feng, A.; Xu, D.; Zhu, Q.; Mao, Y.; Zhao, Y.; Lv, Y.; Han, C.; Liu, R.; et al. Methotrexate, doxorubicin, and cisplatinum regimen is still the preferred option for osteosarcoma chemotherapy: A meta-analysis and clinical observation. Medicine 2019, 98, e15582. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, A.J.; Speetjens, F.M.; Dijkstra, P.D.S.; Fiocco, M.; van de Sande, M.A.J.; Bovée, J.V.M.G.; Gelderblom, H. Single-Center Experience with Ifosfamide Monotherapy as Second-Line Treatment of Recurrent/Metastatic Osteosarcoma. Oncologist 2020, 25, e716–e721. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. Cancer metabolism pipeline breaks new ground. Nat. Rev. Drug Discov. 2016, 15, 735–737. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves first-in-class cancer metabolism drug. Nat. Rev. Drug Discov. 2017, 16, 593. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Bosc, C.; Selak, M.A.; Sarry, J.E. Resistance is futile: Targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment. Cell Metab. 2017, 26, 705–707. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Gallardo, C.; Simón, C. Cells, stem cells, and cancer stem cells. Semin. Reprod. Med. 2013, 31, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Odoux, C.; Fohrer, H.; Hoppo, T.; Guzik, L.; Stolz, D.B.; Lewis, D.W.; Gollin, S.M.; Gamblin, T.C.; Geller, D.A.; Lagasse, E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008, 68, 6932–6941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, V.; Reed-Newman, T.C.; Arnold, L.; Thomas, S.M.; Anant, S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front. Oncol. 2018, 8, 203. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers 2019, 11, 310. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, R.; Santulli, A.; Ferrante, R.D.; Giuliano, M.; De Blasio, A.; Messina, C.; Pirozzi, G.; Tirino, V.; Tesoriere, G.; Vento, R. Identification and expansion of human osteosarcoma-cancer-stem cells by long-term 3-aminobenzamide treatment. J. Cell. Physiol. 2009, 219, 301–313. [Google Scholar] [CrossRef]
- Di Fiore, R.; Fanale, D.; Drago-Ferrante, R.; Chiaradonna, F.; Giuliano, M.; De Blasio, A.; Amodeo, V.; Corsini, L.R.; Bazan, V.; Tesoriere, G.; et al. Genetic and molecular characterization of the human osteosarcoma 3AB-OS cancer stem cell line: A possible model for studying osteosarcoma origin and stemness. J. Cell. Physiol. 2013, 228, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, R.; Guercio, A.; Puleio, R.; Di Marco, P.; Drago-Ferrante, R.; D’Anneo, A.; De Blasio, A.; Carlisi, D.; Di Bella, S.; Pentimalli, F.; et al. Modeling human osteosarcoma in mice through 3AB-OS cancer stem cell xenografts. J. Cell. Biochem. 2012, 113, 3380–3392. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef]
- Tataranni, T.; Agriesti, F.; Ruggieri, V.; Mazzoccoli, C.; Simeon, V.; Laurenzana, I.; Scrima, R.; Pazienza, V.; Capitanio, N.; Piccoli, C. Rewiring carbohydrate catabolism differentially affects survival of pancreatic cancer cell lines with diverse metabolic profiles. Oncotarget 2017, 8, 41265–41281. [Google Scholar] [CrossRef] [Green Version]
- Teta, R.; Della Sala, G.; Esposito, G.; Via, C.W.; Mazzoccoli, C.; Piccoli, C.; Bertin, M.J.; Costantino, V.; Mangoni, A. A joint molecular networking study of a Smenospongia sponge and a cyanobacterial bloom revealed new antiproliferative chlorinated polyketides. Org. Chem. Front. 2019, 6, 1762–1774. [Google Scholar] [CrossRef]
- Seo, B.J.; Yoon, S.H.; Do, J.T. Mitochondrial Dynamics in Stem Cells and Differentiation. Int. J. Mol. Sci. 2018, 19, 3893. [Google Scholar] [CrossRef] [Green Version]
- Movahed, Z.G.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef]
- Fritz, V.; Fajas, L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 2010, 29, 4369–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Anil Kumar, K.; Reddanna, P. Did we get pasteur, warburg, and crabtree on a right note? Front. Oncol. 2013, 3, 186. [Google Scholar] [CrossRef] [Green Version]
- Hammad, N.; Rosas-Lemus, M.; Uribe-Carvajal, S.; Rigoulet, M.; Devin, A. The Crabtree and Warburg effects: Do metabolite-induced regulations participate in their induction? Biochim. Biophys. Acta 2016, 1857, 1139–1146. [Google Scholar] [CrossRef]
- Ling, Y.H.; Consoli, U.; Tornos, C.; Andreeff, M.; Perez-Soler, R. Accumulation of cyclin B1, activation of cyclin B1-dependent kinase and induction of programmed cell death in human epidermoid carcinoma KB cells treated with taxol. Int. J. Cancer 1998, 75, 925–932. [Google Scholar] [CrossRef]
- Barroso-González, J.; Thomas, G. Endosome traffic machinery meets the p53-p21 axis. Mol. Cell. Oncol. 2015, 2, e975075. [Google Scholar] [CrossRef] [PubMed]
- Gottifredi, V.; McKinney, K.; Poyurovsky, M.V.; Prives, C. Decreased p21 levels are required for efficient restart of DNA synthesis after S phase block. J. Biol. Chem. 2004, 279, 5802–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Nishikawa, M.; Haque, A.M.; Hirose, M.; Mashimo, M.; Sato, E.; Inoue, M. Mitochondrial density determines the cellular sensitivity to cisplatin-induced cell death. Am. J. Physiol. Cell Physiol. 2005, 289, C1466–C1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731. [Google Scholar] [CrossRef] [Green Version]
- Vacanti, N.M.; Metallo, C.M. Exploring metabolic pathways that contribute to the stem cell phenotype. Biochim. Biophys. Acta 2013, 1830, 2361–2369. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.F.; Zhang, C.; Wu, Y.Q.; Zhuang, G.; Ye, Z.; Zhang, C.S.; Lin, S.C. Glutaminase GLS1 senses glutamine availability in a non-enzymatic manner triggering mitochondrial fusion. Cell Res. 2018, 28, 865–867. [Google Scholar] [CrossRef] [Green Version]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar]
- Cocetta, V.; Ragazzi, E.; Montopoli, M. Mitochondrial Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2019, 20, 3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, A.; Tsunoda, Y.; Furuya, K.; Oyamada, H.; Tsuji, M.; Udaka, Y.; Hosonuma, M.; Shirako, H.; Ichimura, N.; Kiuchi, Y. Cancer stem-like cells have cisplatin resistance and miR-93 regulate p21 expression in breast cancer. Cancer Transl. Med. 2018, 4, 48–53. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ye, Z.; Lai, W.; Rao, J.; Huang, W.; Zhang, X.; Yao, Z.; Lou, T. Activation of Sirtuin 3 by Silybin Attenuates Mitochondrial Dysfunction in Cisplatin-induced Acute Kidney Injury. Front. Pharmacol. 2017, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Deng, C.X.; Finley, L.W.; Kim, H.S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Sirtuins in aging and age-related diseases. In Handbook of the Biology of Aging, 7th ed.; Masoro, E.J., Austad, S.N., Eds.; Academic Press: Cambridge, MA, USA, 2011; Chapter 11; pp. 243–274. [Google Scholar]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lettieri-Barbato, D.; Aquilano, K. Pushing the Limits of Cancer Therapy: The Nutrient Game. Front. Oncol. 2018, 8, 148. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Sala, G.; Pacelli, C.; Agriesti, F.; Laurenzana, I.; Tucci, F.; Tamma, M.; Capitanio, N.; Piccoli, C. Unveiling Metabolic Vulnerability and Plasticity of Human Osteosarcoma Stem and Differentiated Cells to Improve Cancer Therapy. Biomedicines 2022, 10, 28. https://doi.org/10.3390/biomedicines10010028
Della Sala G, Pacelli C, Agriesti F, Laurenzana I, Tucci F, Tamma M, Capitanio N, Piccoli C. Unveiling Metabolic Vulnerability and Plasticity of Human Osteosarcoma Stem and Differentiated Cells to Improve Cancer Therapy. Biomedicines. 2022; 10(1):28. https://doi.org/10.3390/biomedicines10010028
Chicago/Turabian StyleDella Sala, Gerardo, Consiglia Pacelli, Francesca Agriesti, Ilaria Laurenzana, Francesco Tucci, Mirko Tamma, Nazzareno Capitanio, and Claudia Piccoli. 2022. "Unveiling Metabolic Vulnerability and Plasticity of Human Osteosarcoma Stem and Differentiated Cells to Improve Cancer Therapy" Biomedicines 10, no. 1: 28. https://doi.org/10.3390/biomedicines10010028
APA StyleDella Sala, G., Pacelli, C., Agriesti, F., Laurenzana, I., Tucci, F., Tamma, M., Capitanio, N., & Piccoli, C. (2022). Unveiling Metabolic Vulnerability and Plasticity of Human Osteosarcoma Stem and Differentiated Cells to Improve Cancer Therapy. Biomedicines, 10(1), 28. https://doi.org/10.3390/biomedicines10010028