
Phenotypic Heterogeneity among GBA p.R202X Carriers in Lewy Body Spectrum Disorders


 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Standard Protocol Approvals, Registrations, and Patient Consents
2.2. Genetic Analysis
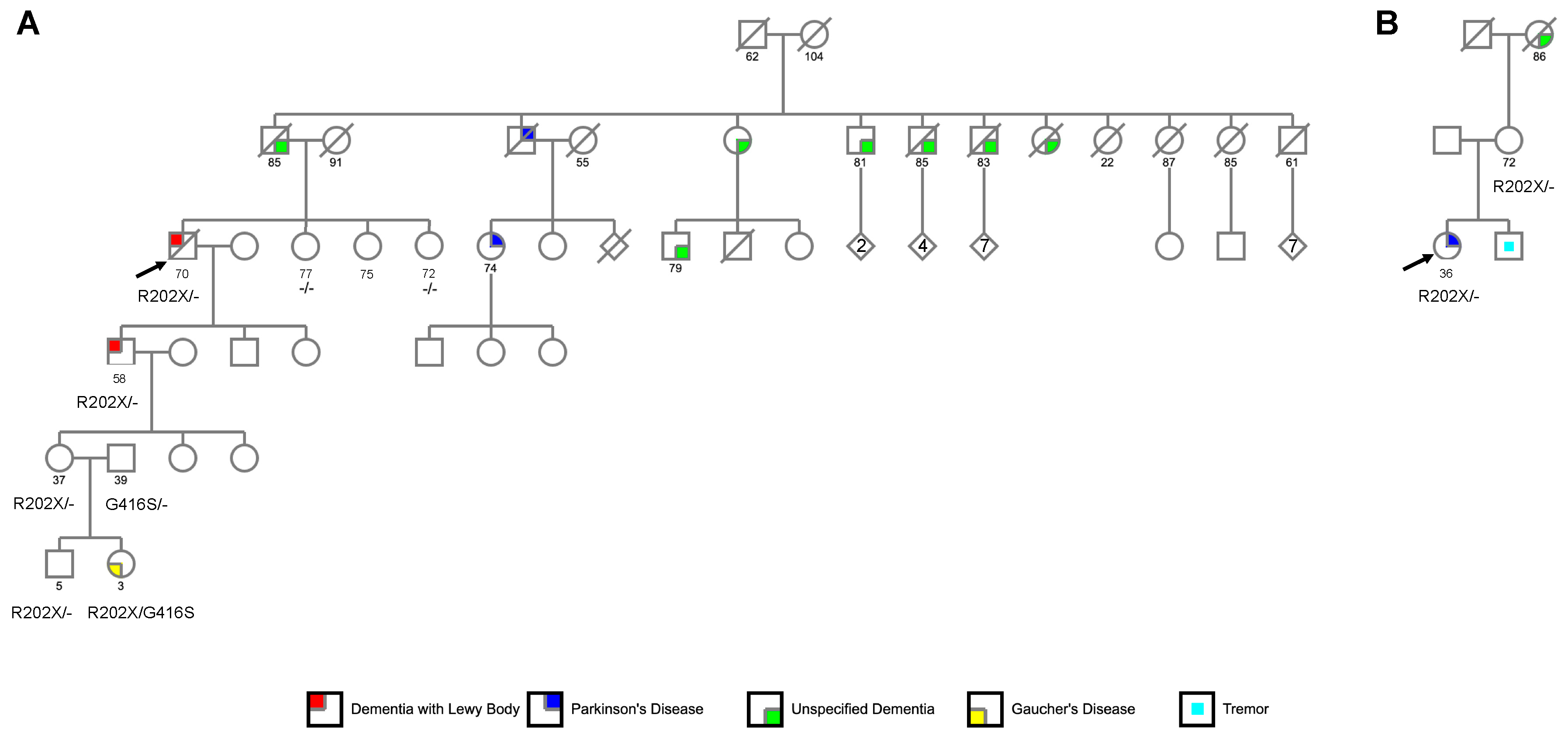
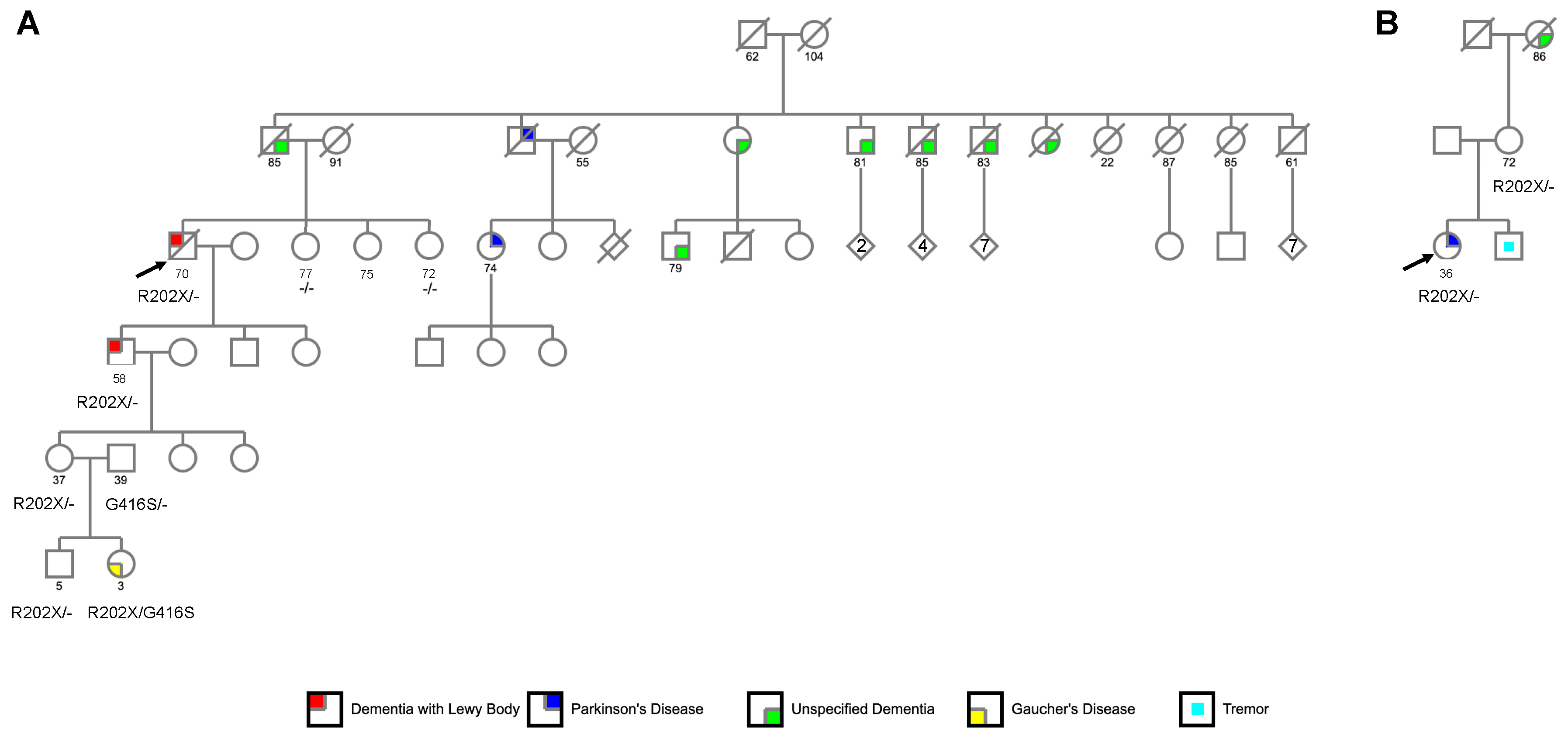
2.2.1. Family 1
2.2.2. Family 2
2.3. Literature and Database Search
3. Results
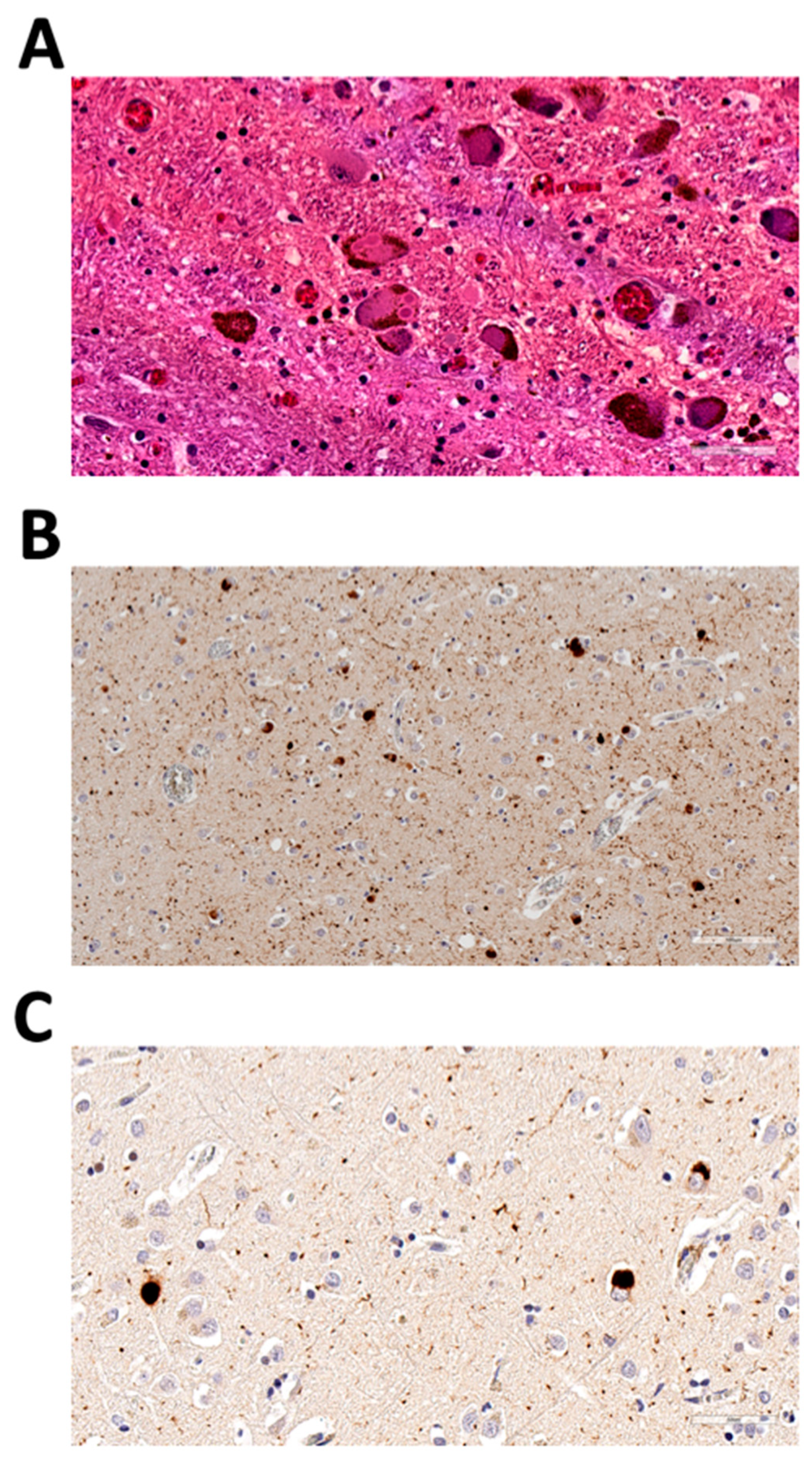
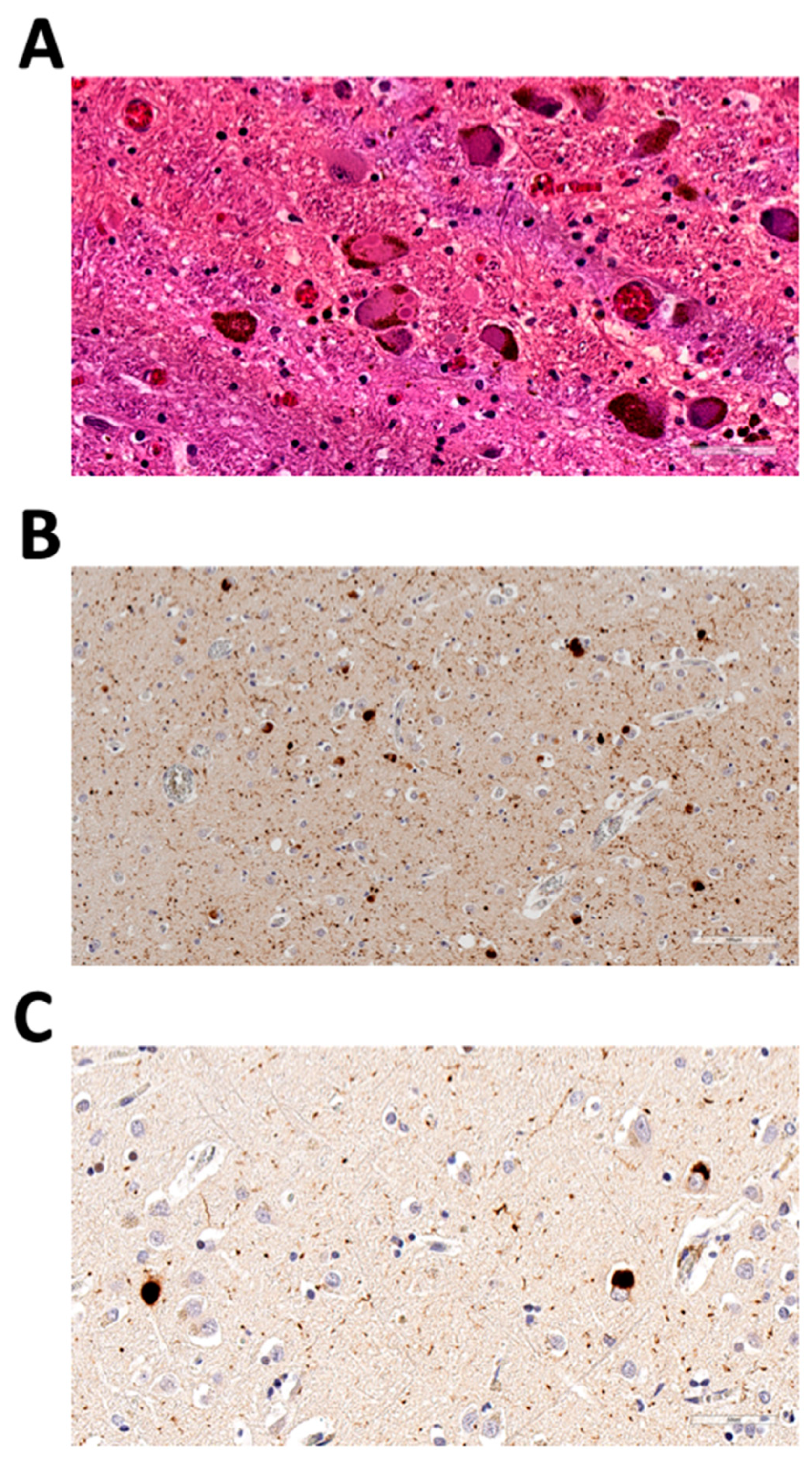
3.1. Case Presentation
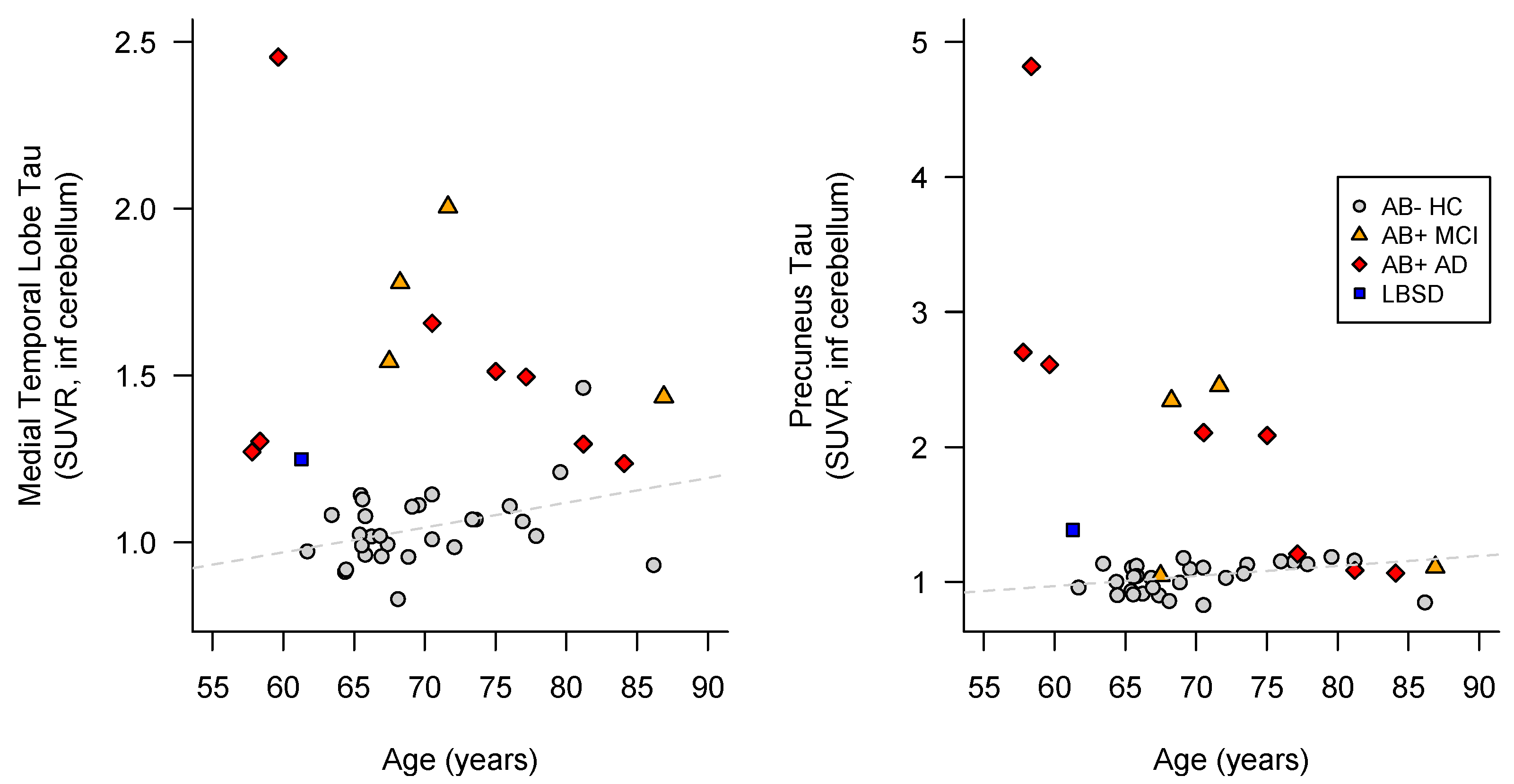
3.1.1. Family 1
3.1.2. Family 2
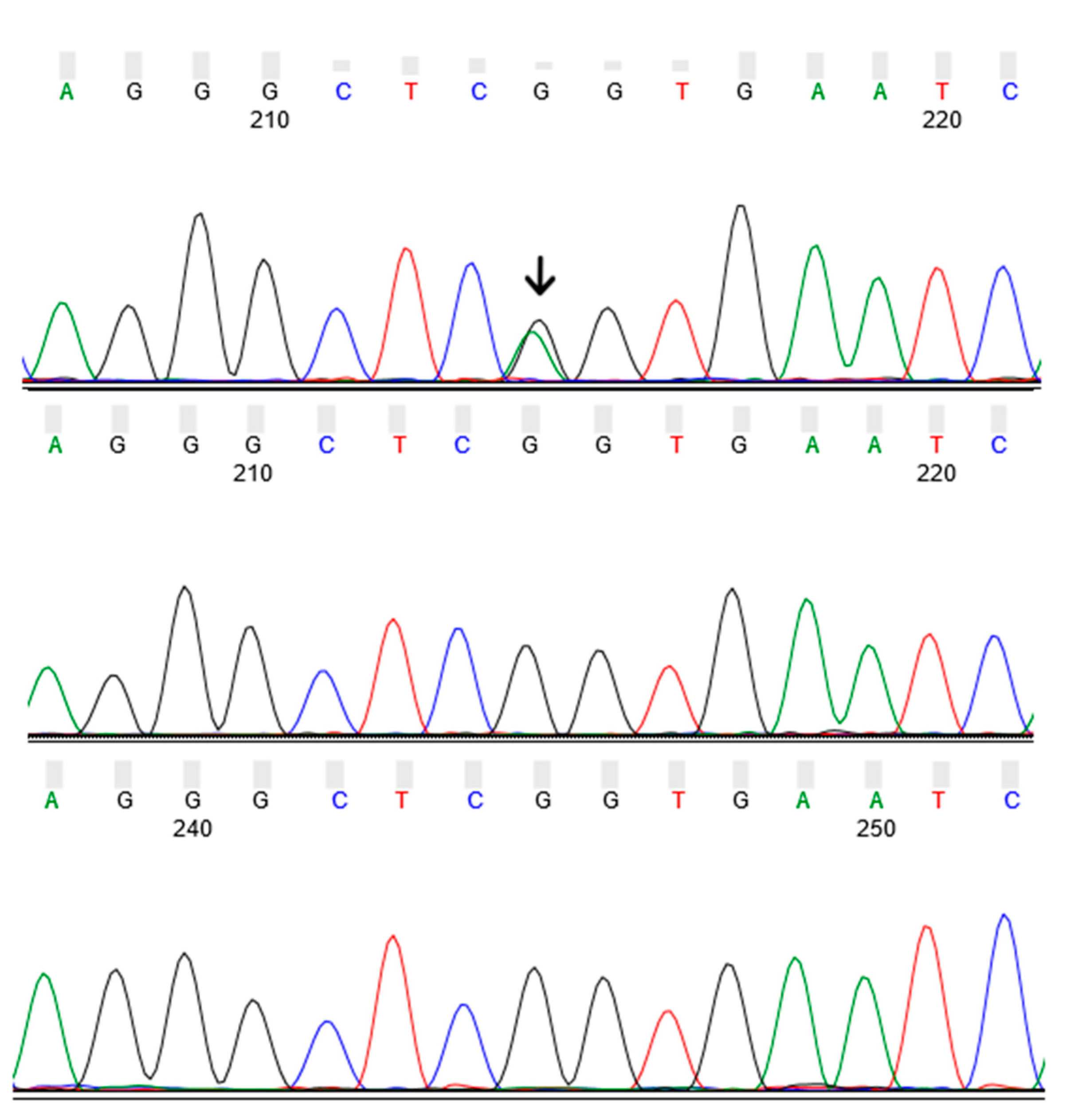
3.2. Identification of GBA p.R202X Variant
3.3. Literature Search of GBA p.R202X Variant Carriers
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Irwin, D.J.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Duda, J.E.; Xie, S.X.; Lee, E.B.; Van Deerlin, V.M.; Lopez, O.L.; Kofler, J.K.; et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 2017, 16, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Riboldi, G.M.; Di Fonzo, A.B. Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Shu, L.; Sun, Q.; Zhou, X.; Pan, H.; Guo, J.; Tang, B. Integrated Genetic Analysis of Racial Differences of Common GBA Variants in Parkinson’s Disease: A Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Pastores, G.M.; Hughes, D.A. Gaucher disease. In GeneReviews™; Pagon, R.A., Adam, M.P., Bird, T.D., Dolan, C.R., Fong, C.T., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 2000. [Google Scholar]
- Guo, Y.; Ding, X.; Shen, Y.; Lyon, G.J.; Wang, K. SeqMule: Automated pipeline for analysis of human exome/genome sequencing data. Sci. Rep. 2015, 5, 14283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beecham, G.W.; Bis, J.; Martin, E.; Choi, S.-H.; DeStefano, A.L.; van Duijn, C.; Fornage, M.; Gabriel, S.; Koboldt, D.; Larson, D.; et al. The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol. Genet. 2017, 3, e194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finckh, U.; Seeman, P.; Von Widdern, O.C.; Rolfs, A. Simple PCR amplification of the entire glucocerebrosidase gene (GBA) coding region for diagnostic sequence analysis. DNA Seq. 1998, 8, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Tayebi, N.; Stubblefield, B.K.; Park, J.; Orvisky, E.; Walker, J.M.; LaMarca, M.E.; Sidransky, E. Reciprocal and Nonreciprocal Recombination at the Glucocerebrosidase Gene Region: Implications for Complexity in Gaucher Disease. Am. J. Hum. Genet. 2003, 72, 519–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef]
- Guerreiro, R.; Sassi, C.; Gibbs, J.R.; Edsall, C.; Hernandez, D.; Brown, K.; Lupton, M.K.; Parkinnen, L.; Ansorge, O.; Hodges, A.; et al. A comprehensive assessment of benign genetic variability for neurodegenerative disorders. bioRxiv 2018, 270686. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.P.; Berg, L.; Danziger, W.L.; Coben, L.A.; Martin, R.L. A New Clinical Scale for the Staging of Dementia. Br. J. Psychiatry 1982, 140, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool For Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.F.; Leverenz, J.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Do, S.A.F.; Wood-Siverio, C.; et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Chahine, L.M.; Qiang, J.; Ashbridge, E.; Minger, J.; Yearout, D.; Horn, S.; Colcher, A.; Hurtig, H.I.; Lee, V.M.-Y.; Van Deerlin, V.M.; et al. Clinical and Biochemical Differences in Patients Having Parkinson Disease with vs WithoutGBAMutations. JAMA Neurol. 2013, 70, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism with Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef] [Green Version]
- Barrett, M.J.; Giraldo, P.; Capablo, J.L.; Alfonso, P.; Irun, P.; Garcia-Rodriguez, B.; Pocovi, M.; Pastores, G.M. Greater risk of parkinsonism associated with non-N370S GBA1 mutations. J. Inherit. Metab. Dis. 2013, 36, 575–580. [Google Scholar] [CrossRef] [Green Version]
- Tsuang, D.; Leverenz, J.B.; Lopez, O.L.; Hamilton, R.L.; Bennett, D.A.; Schneider, J.A.; Buchman, A.S.; Larson, E.B.; Crane, P.K.; Kaye, J.A.; et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013, 70, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Levstek, T.; Redenšek, S.; Trošt, M.; Dolžan, V.; Podkrajšek, K. Assessment of the Telomere Length and Its Effect on the Symptomatology of Parkinson’s Disease. Antioxidants 2021, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Tropea, T.F.; Mak, J.; Guo, M.H.; Xie, S.X.; Suh, E.; Rick, J.; Siderowf, A.; Weintraub, D.; Grossman, M.; Irwin, D.; et al. TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann. Neurol. 2019, 85, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, F.; Marin-Lahoz, J.; Martinez-Horta, S.; Pagonabarraga, J.; Kulisevsky, J. Cortical Thinning Associated with Age and CSF Biomarkers in Early Parkinson’s Disease is Modified by the SCNA rs356181 Polymorphism. Neurodegener. Dis. 2018, 18, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Ushakova, A.; Doitsidou, M.; Tzoulis, C.; Tysnes, O.-B.; Dalen, I.; Pedersen, K.F.; Alves, G.; Maple-Grødem, J. The impact of common genetic variants in cognitive decline in the first seven years of Parkinson’s disease: A longitudinal observational study. Neurosci. Lett. 2021, 764, 136243. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Prada-Dacasa, P.; Santos, C.; Adame-Castillo, C.; Campdelacreu, J.; Reñé, R.; Gascón-Bayarri, J.; Ispierto, L.; Álvarez, R.; Ariza, A.; et al. GBA Mutations Are Associated with Earlier Onset and Male Sex in Dementia with Lewy Bodies. Mov. Disord. 2016, 31, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Swan, M.; Doan, N.; Ortega, R.A.; Barrett, M.; Nichols, W.; Ozelius, L.; Soto-Valencia, J.; Boschung, S.; Deik, A.; Sarva, H.; et al. Neuropsychiatric characteristics of GBA-associated Parkinson disease. J. Neurol. Sci. 2016, 370, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, S.; Cattarossi, S.; Bembi, B.; Dardis, A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J. Mol. Diagn. 2017, 19, 733–741. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Diagnosis | Sex | Age-at-Onset | Age-at-Death or Last Assessed Normal | Autopsy-Confirmed | APOE Genotype | Ancestry |
|---|---|---|---|---|---|---|---|
| Present—Family 1 | LBD | M | 70 | 74 | Y | 3/3 | Irish-American |
| Present—Family 1 | LBSD | M | 58 | - | - | 3/4 | Irish-American |
| Present—Family 2 | PD | F | 36 | - | - | 3/3 | British-American |
| Present—Family 2 | Unaffected | F | - | 72 | - | - | British-American |
| Irwin, D.J., et al. [1] | LBSD | M | 50 | 57 | Y | 3/3 | NA |
| Mata, I.F., et al.[16], Sidransky, E., et al. [17], Chahine, L.M., et al. [18], Davis, M.Y., et al. [19] | PD | F | 47 | - | - | - | African-American |
| Barrett, M.J., et al. [20] | - | - | - | - | - | - | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Napolioni, V.; Fredericks, C.A.; Kim, Y.; Channappa, D.; Khan, R.R.; Kim, L.H.; Zafar, F.; Couthouis, J.; Davidzon, G.A.; Mormino, E.C.; et al. Phenotypic Heterogeneity among GBA p.R202X Carriers in Lewy Body Spectrum Disorders. Biomedicines 2022, 10, 160. https://doi.org/10.3390/biomedicines10010160
Napolioni V, Fredericks CA, Kim Y, Channappa D, Khan RR, Kim LH, Zafar F, Couthouis J, Davidzon GA, Mormino EC, et al. Phenotypic Heterogeneity among GBA p.R202X Carriers in Lewy Body Spectrum Disorders. Biomedicines. 2022; 10(1):160. https://doi.org/10.3390/biomedicines10010160
Chicago/Turabian StyleNapolioni, Valerio, Carolyn A. Fredericks, Yongha Kim, Divya Channappa, Raiyan R. Khan, Lily H. Kim, Faria Zafar, Julien Couthouis, Guido A. Davidzon, Elizabeth C. Mormino, and et al. 2022. "Phenotypic Heterogeneity among GBA p.R202X Carriers in Lewy Body Spectrum Disorders" Biomedicines 10, no. 1: 160. https://doi.org/10.3390/biomedicines10010160
APA StyleNapolioni, V., Fredericks, C. A., Kim, Y., Channappa, D., Khan, R. R., Kim, L. H., Zafar, F., Couthouis, J., Davidzon, G. A., Mormino, E. C., Gitler, A. D., Montine, T. J., Schüle, B., & Greicius, M. D. (2022). Phenotypic Heterogeneity among GBA p.R202X Carriers in Lewy Body Spectrum Disorders. Biomedicines, 10(1), 160. https://doi.org/10.3390/biomedicines10010160