
A High-Response Electrochemical As(III) Sensor Using Fe3O4–rGO Nanocomposite Materials




Abstract
1. Introduction
2. Experimental
2.1. Materials/Chemicals
2.2. Instrumentation
2.3. Synthesis of Fe3O4–rGO Nanocomposites
2.4. Preparation of the Fe3O4–rGO-Nanocomposite-Modified Electrode
2.5. Electrochemical Measurements
3. Results and Discussion
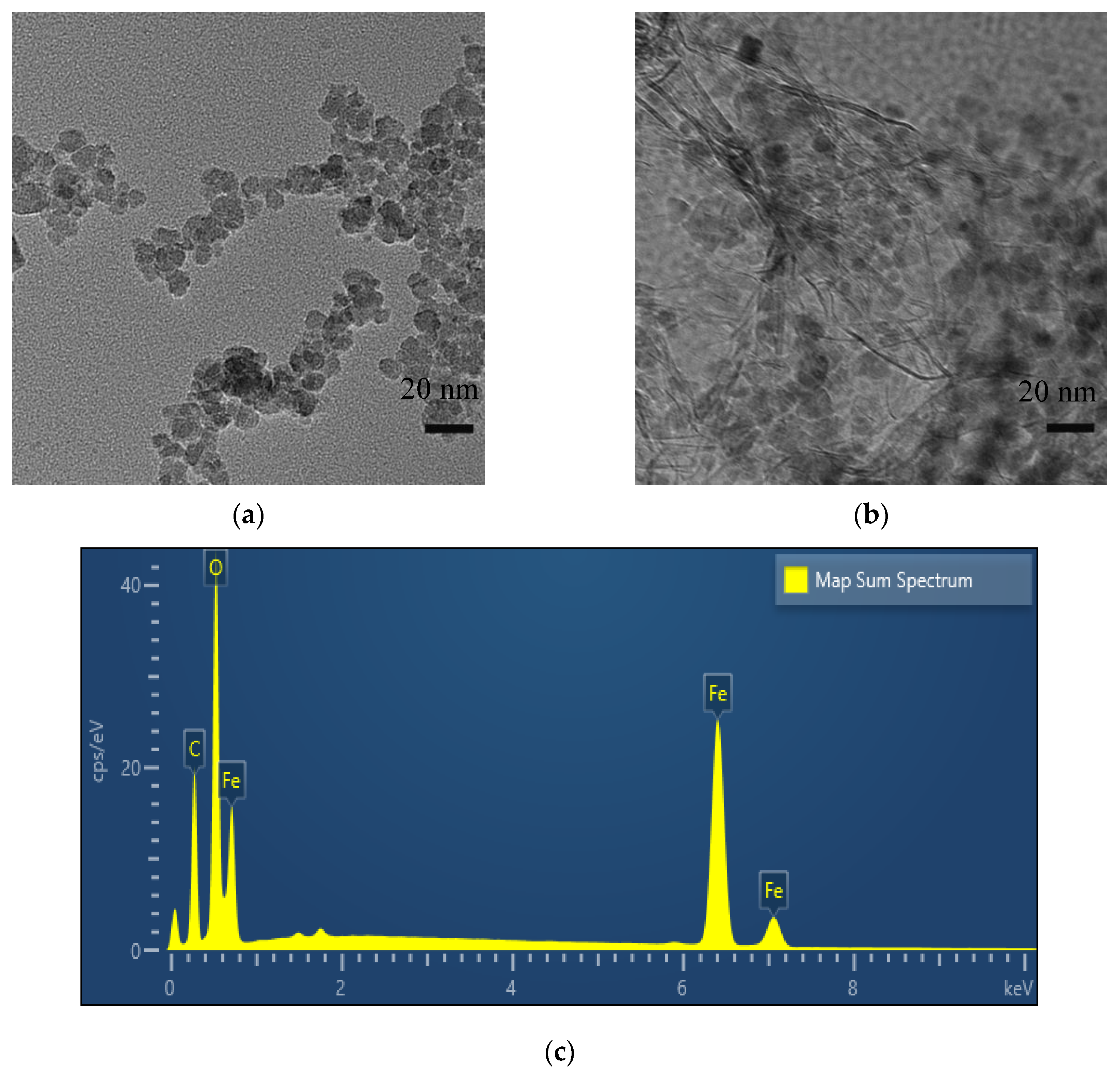
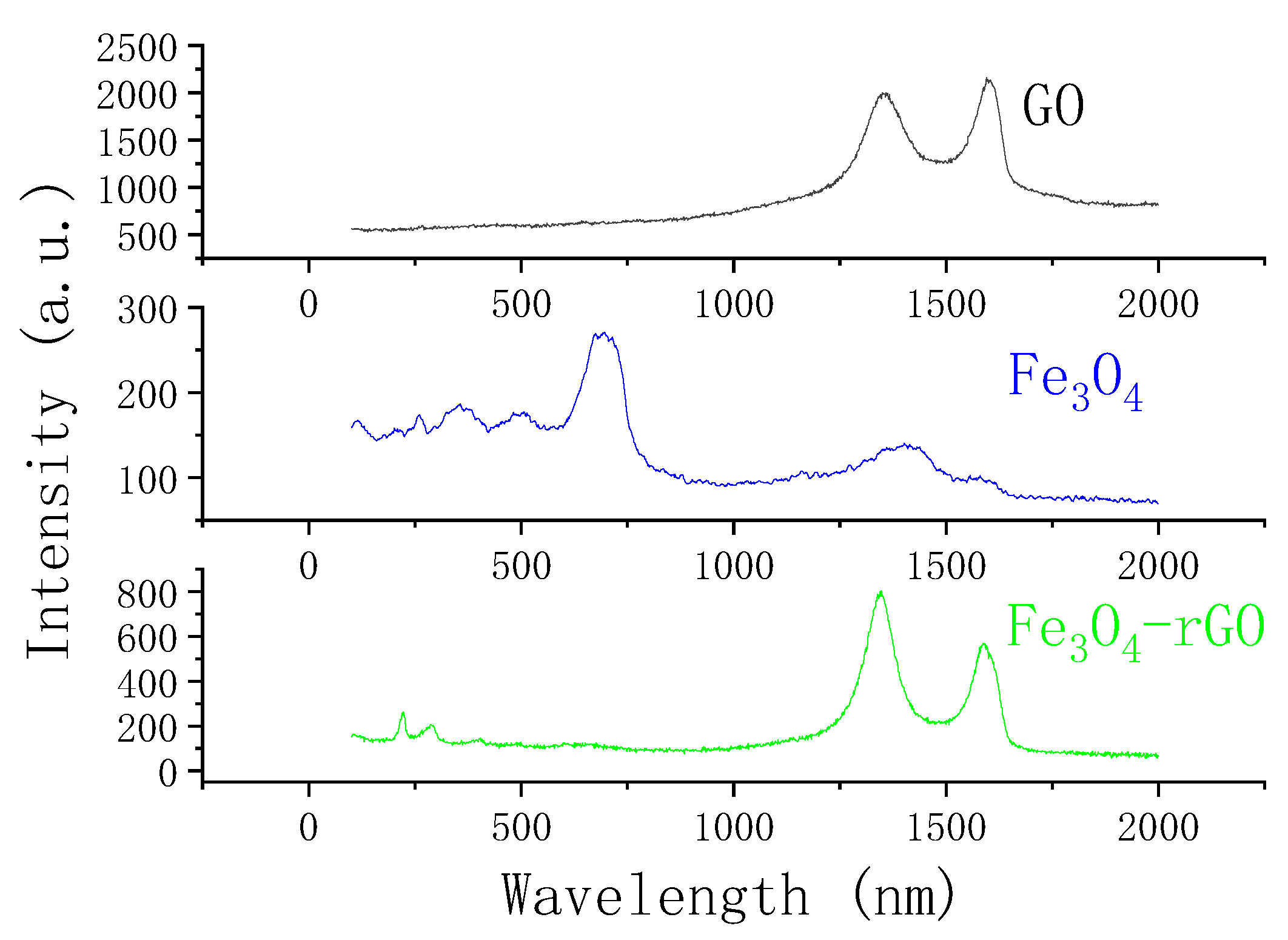
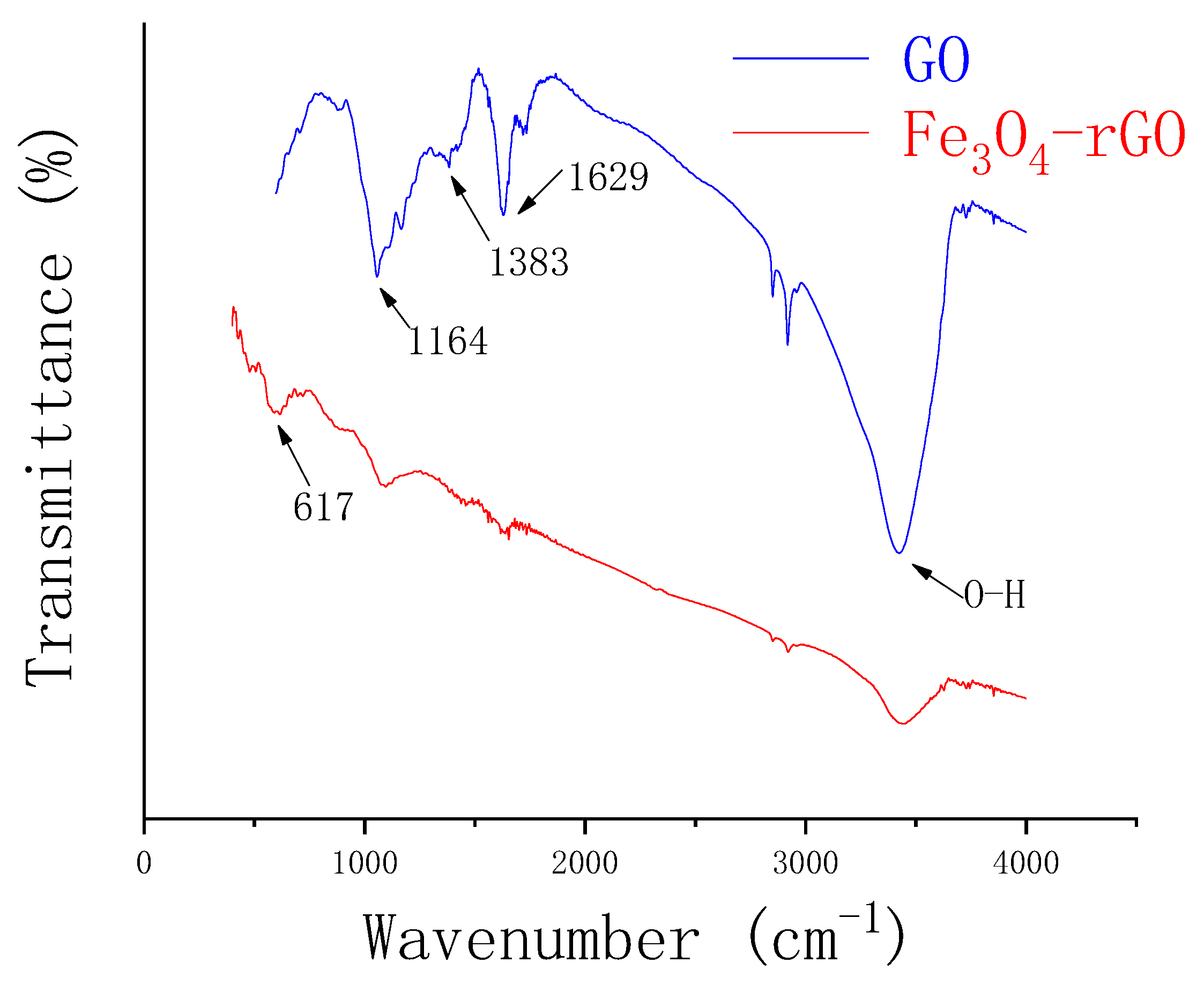
3.1. Characterization of Fe3O4–rGO Nanocomposites
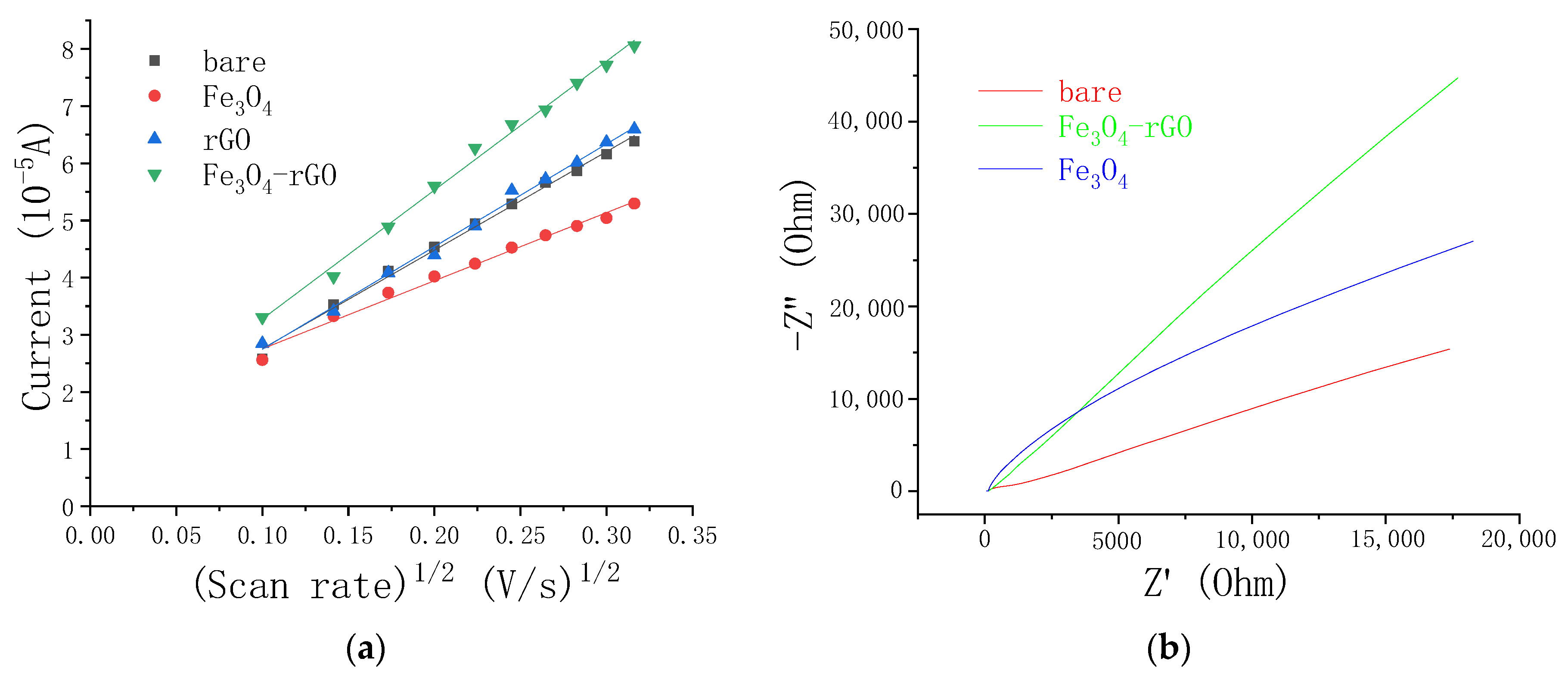
3.2. Electrochemical Characterization of NC/GC Electrodes
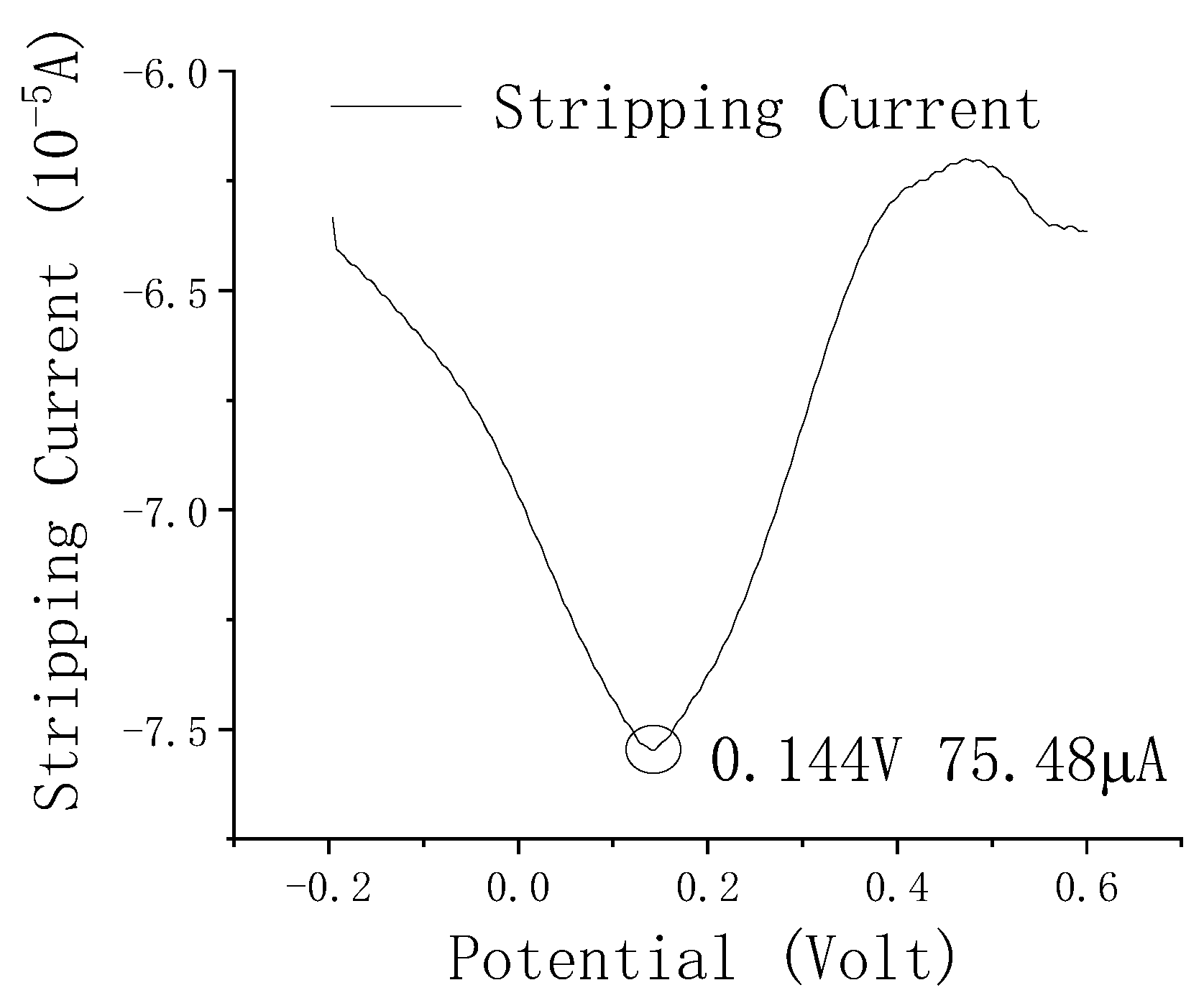
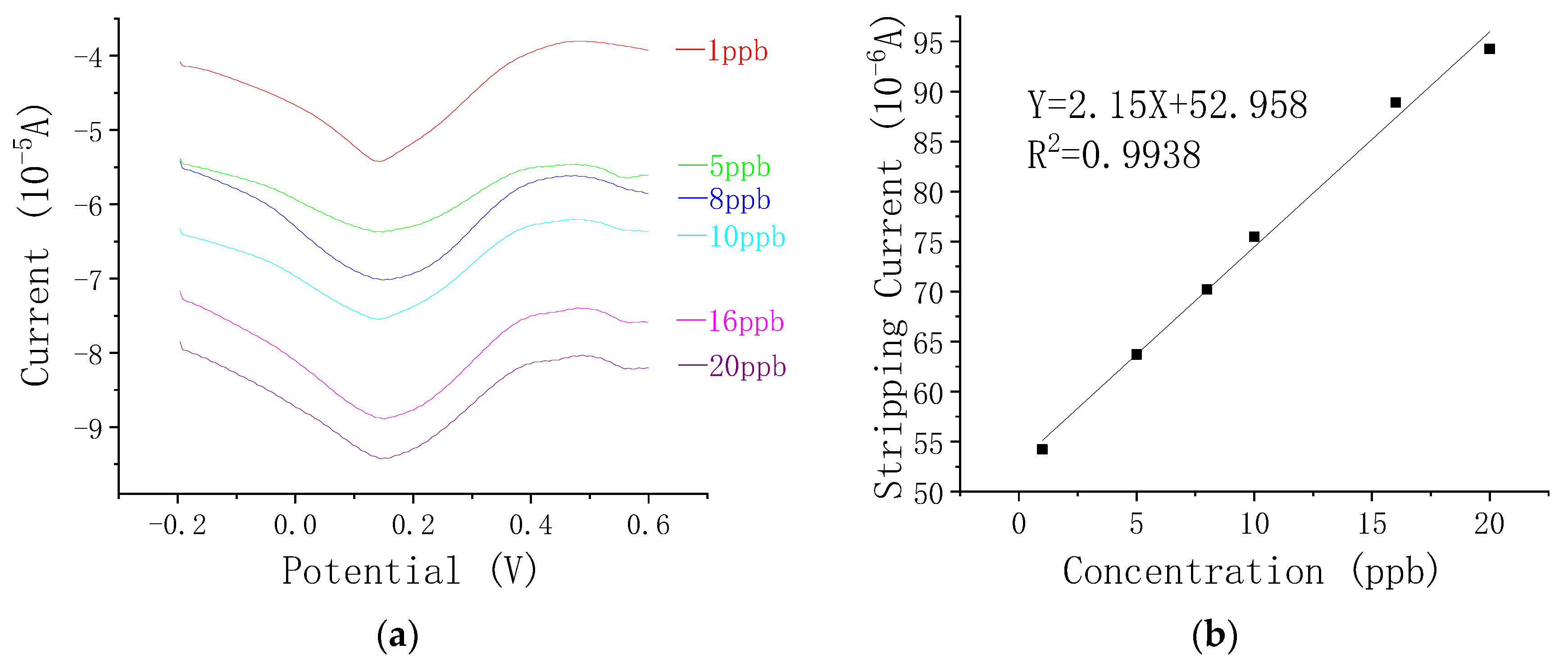
3.3. Analytical Performance of the Fe3O4–rGO-Nanocomposite—Modified Electrode towards As(III)
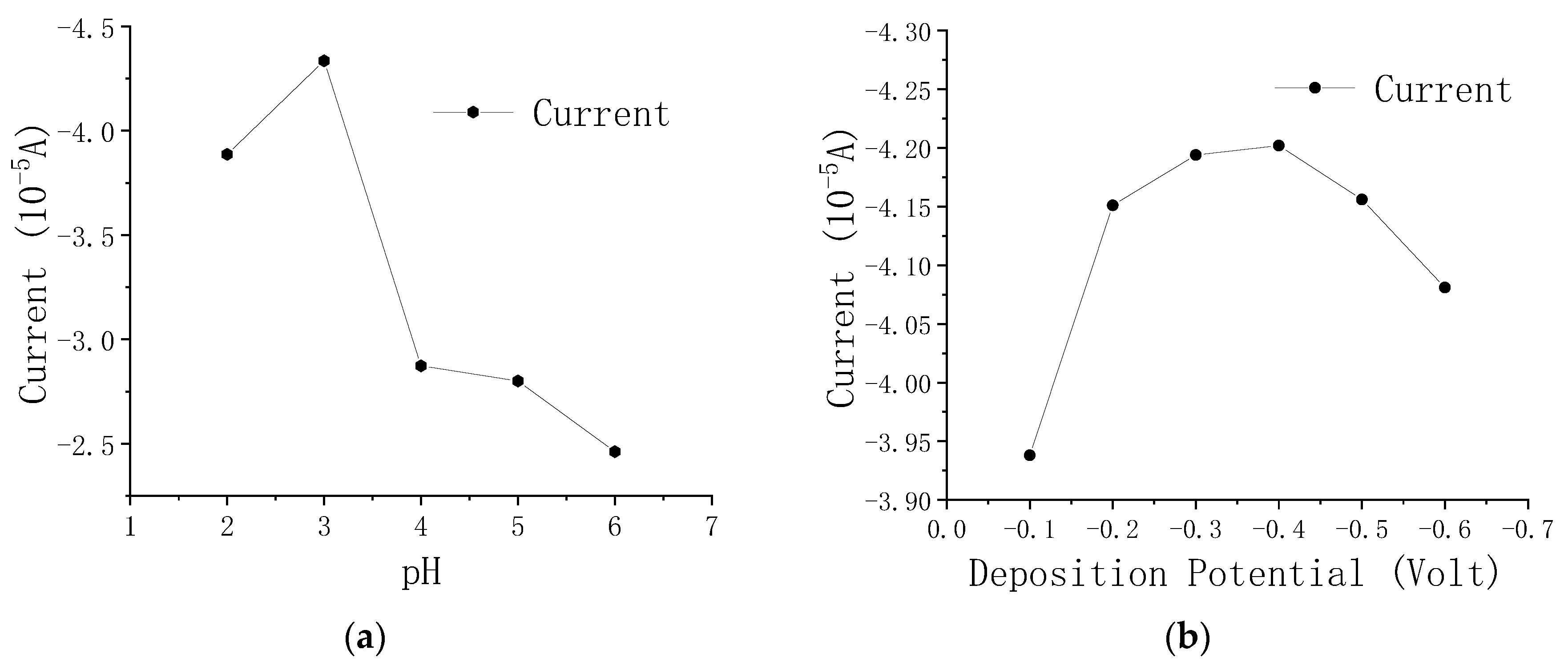
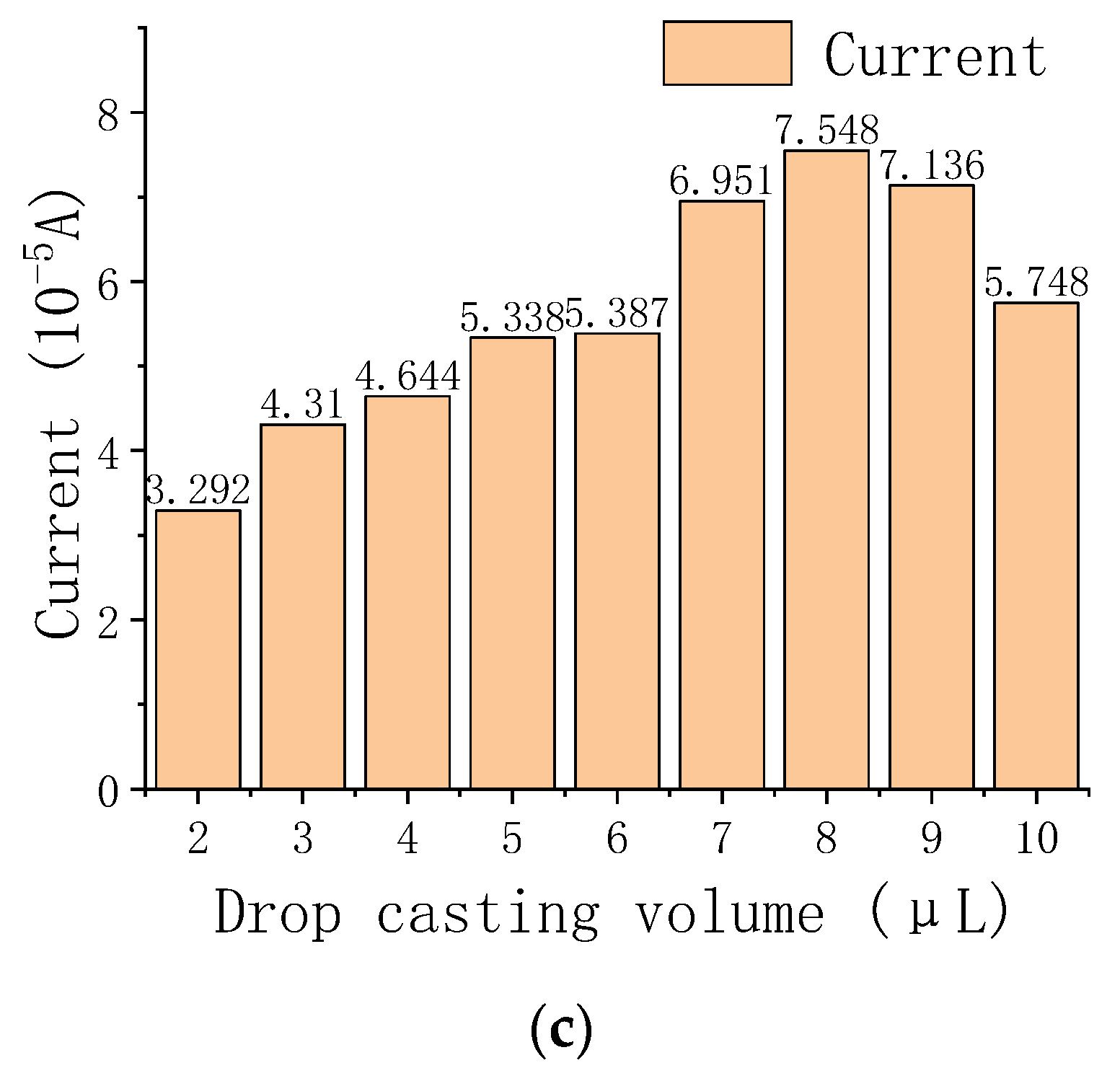
3.4. Optimization of Experimental Conditions for the Electrochemical Detection of As(III)
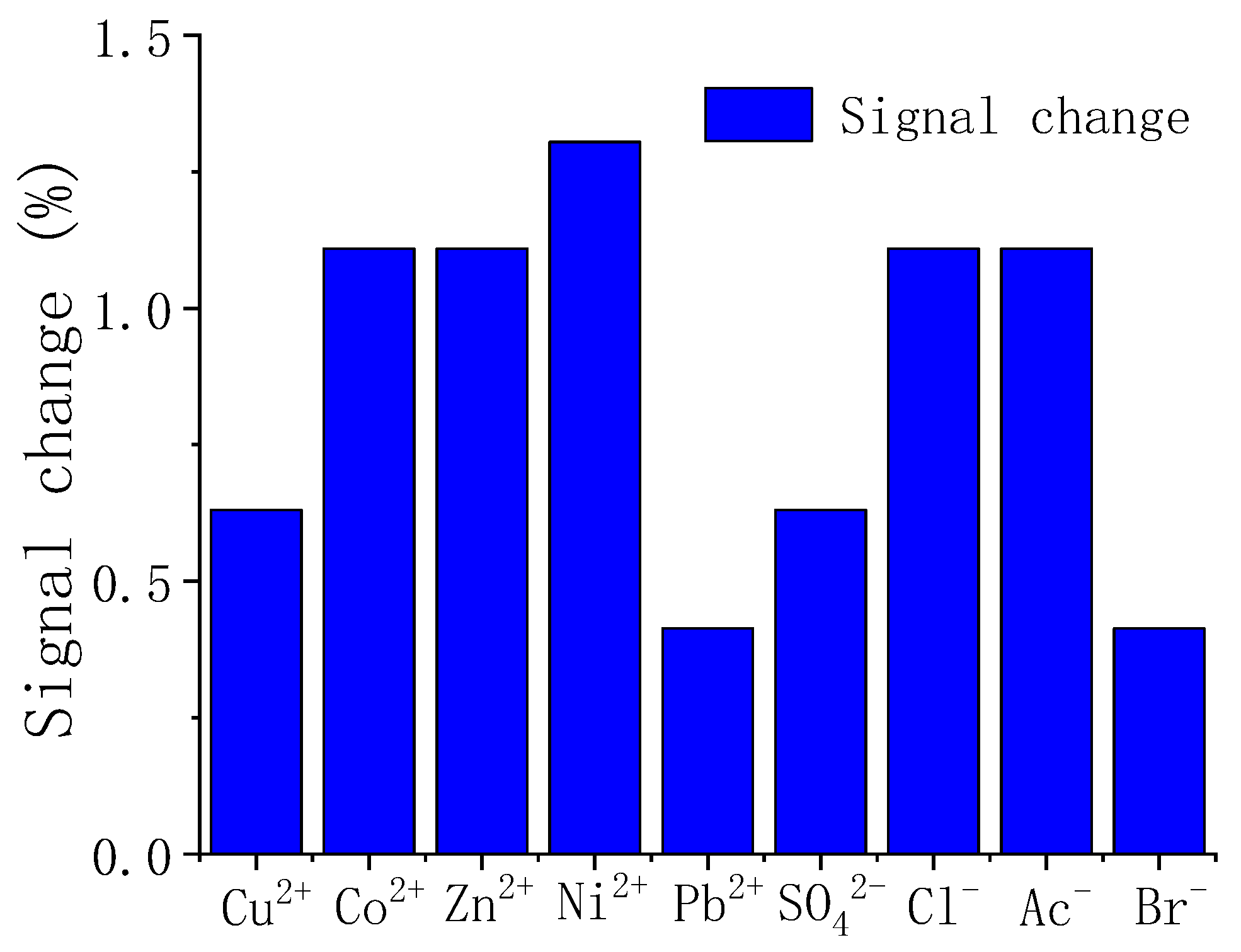
3.5. Interference Measurement
3.6. Stability and Reproducibility Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Flavio, M.; Maria, L.; Antonio, R. Heavy Metal/Toxins Detection Using Electronic Tongues. Chemosensors 2019, 7, 36. [Google Scholar]
- Liu, Z.-G.; Huang, X.-J. Voltammetric determination of inorganic arsenic. TrAC Trends Anal. Chem. 2014, 60, 25–35. [Google Scholar] [CrossRef]
- Annija, L.; Jonh, C. A Rewiew of Microfluidic Detection Strategies for Heavy Metals in Water. Chemosensors 2021, 9, 60. [Google Scholar]
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef]
- Stojanovic, R.S.; Bond, A.M.; Butler, E.C.V. Liquid chromatography-electrochemical detection of inorganic arsenic using a wall jet cell with conventional and microsized platinum disk electrodes. Anal. Chem. 1990, 62, 2692–2697. [Google Scholar] [CrossRef]
- Simm, A.O.; Banks, C.E.; Compton, R.G. Sonically assisted electroanalytical detection of ultratrace arsenic. Anal. Chem. 2004, 76, 5051–5055. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Srivastava, R.C.; Seth, P.K. Direct Estimation of Total Arsenic Using a Novel Metal Side Disk Rotating Electrode. Electroanalysis 2003, 15, 1410–1414. [Google Scholar] [CrossRef]
- March, G.; Nguyen, T.D.; Piro, B. Modified electrodes used for electrochemical detection of metal ions in environmental analysis. Biosensors (Basel) 2015, 5, 241–275. [Google Scholar] [CrossRef] [PubMed]
- Ratinac, K.R.; Yang, W.; Gooding, J.J.; Thordarson, P.; Braet, F. Graphene and Related Materials in Electrochemical Sensing. Electroanalysis 2011, 23, 803–826. [Google Scholar] [CrossRef]
- Aragay, G.; Merkoçi, A. Nanomaterials application in electrochemical detection of heavy metals. Electrochim. Acta 2012, 84, 49–61. [Google Scholar] [CrossRef]
- Aikens, D.A. Electrochemical methods, fundamentals and applications. J. Chem. Educ. 1983, 60, A25. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, N.; Wang, H.; Liang, Q.; Bian, Z. Pd-Ag/Graphene Electrochemical Sensor for Chlorophenol Contaminant Determination. J. Electrochem. Soc. 2019, 166, B266–B275. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Li, H.; Wang, G.; Long, Y.; Li, X.; Yang, B. In Situ Detection of Melatonin and Pyridoxine in Plants Using a CuO-Poly(L-lysine)/Graphene-Based Electrochemical Sensor. ACS Sustain. Chem. Eng. 2019, 7, 19537–19545. [Google Scholar] [CrossRef]
- Li, F.; Wang, A.; Lai, G.; Su, W.; Malherbe, F.; Yu, J.; Lin, C.; Yu, A. Defects regulating of graphene ink for electrochemical determination of ascorbic acid, dopamine and uric acid. Talanta 2018, 180, 248–253. [Google Scholar]
- Ramesha, G.K.; Sampath, S. In-situ formation of graphene–lead oxide composite and its use in trace arsenic detection. Sens. Actuators B Chem. 2011, 160, 306–311. [Google Scholar] [CrossRef]
- Kempegowda, R.; Antony, D.; Malingappa, P. Graphene–platinum nanocomposite as a sensitive and selective voltammetric sensor for trace level arsenic quantification. Int. J. Smart Nano Mater. 2014, 5, 17–32. [Google Scholar] [CrossRef]
- Khatamian, M.; Khodakarampoor, N.; Saket-Oskoui, M. Efficient removal of arsenic using graphene-zeolite based composites. J. Colloid Interface Sci. 2017, 498, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.A.; Sheng, T.; Sun, C.; Xue, X.; Tan, L.; Xu, X. Arsenic removal from aqueous solutions using Fe3O4-HBC composite: Effect of calcination on adsorbents performance. PLoS ONE 2014, 9, e100704. [Google Scholar] [CrossRef]
- Dixit, S.; Hering, J.G. Comparison of arsenic (V) and arsenic (III) sorption onto iron oxide minerals: Implications for arsenic mobility. Environ. Sci. Technol. 2003, 37, 4182–4189. [Google Scholar] [CrossRef]
- He, H.; Gao, C. Graphene nanosheets decorated with Pd, Pt, Au, and Ag nanoparticles: Synthesis, characterization, and catalysis applications. Sci. China Chem. 2011, 54, 397–404. [Google Scholar] [CrossRef]
- Stoller, M.D.; Park, S.; Zhu, Y.; An, J.; Ruoff, R.S. Graphene-based ultracapacitors. Nano Lett. 2008, 8, 3498–3502. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Ritter, K.; Odziemkowski, M.S.; Gillham, R.W. An in situ study of the role of surface films on granular iron in the permeable iron wall technology. J. Contam. Hydrol. 2002, 55, 87–111. [Google Scholar] [CrossRef]
- Sohouli, E.; Khosrowshahi, E.M.; Radi, P.; Naghian, E.; Nasrabadi, M.R.; Ahmadi, F. Electrochemical sensor based on modified methylcellulose by graphene oxide and Fe3O4 nanoparticles: Application in the analysis of uric acid content in urine. J. Electroanal. Chem. 2020, 877, 114503. [Google Scholar] [CrossRef]
- Chimezie, A.B.; Hajian, R.; Yusof, N.A.; Woi, P.M.; Shams, N. Fabrication of reduced graphene oxide-magnetic nanocomposite (rGO-Fe3O4) as an electrochemical sensor for trace determination of As(III) in water resources. J. Electroanal. Chem. 2017, 796, 33–42. [Google Scholar] [CrossRef]
- Simm, A.O.; Banks, C.E.; Wilkins, S.J.; Karousos, N.G.; Davis, J.; Compton, R.G. A comparison of different types of gold-carbon composite electrode for detection of arsenic (III). Anal. Bioanal. Chem. 2005, 381, 979–985. [Google Scholar] [CrossRef]
- Ali, M.A.; Kamil Reza, K.; Srivastava, S.; Agrawal, V.V.; John, R.; Malhotra, B.D. Lipid-lipid interactions in aminated reduced graphene oxide interface for biosensing application. Langmuir 2014, 30, 4192–4201. [Google Scholar] [CrossRef]
- Mukherjee, M.D.; Dhand, C.; Dwivedi, N.; Singh, B.P.; Sumana, G.; Agarwal, V.V.; Tawale, J.S.; Malhotra, B.D. Facile synthesis of 2-dimensional transparent graphene flakes for nucleic acid detection. Sens. Actuators B Chem. 2015, 210, 281–289. [Google Scholar] [CrossRef]
- Das, M.; Dhand, C.; Sumana, G.; Srivastava, A.K.; Nagarajan, R.; Nain, L.; Iwamoto, M.; Manaka, T.; Malhotra, B.D. Electrophoretic fabrication of chitosan-zirconium-oxide nanobiocomposite platform for nucleic acid detection. Biomacromolecules 2011, 12, 540–547. [Google Scholar] [CrossRef]
- Salinas-Torres, D.; Huerta, F.; Montilla, F.; Morallón, E. Study on elect roactive and electrocatalytic surfaces of single walled carbon nanotube-modified electrodes. Electrochim. Acta 2011, 56, 2464–2470. [Google Scholar] [CrossRef]
- Jia, L.; Wang, H. Electrochemical reduction synthesis of graphene/Nafion nanocomposite film and its performance on the detection of 8-hydroxy-2′-deoxyguanosine in the presence of uric acid. J. Electroanal. Chem. 2013, 705, 37–43. [Google Scholar] [CrossRef]
- Macdonald, J.R.; Barsoukov, E. Impedance Spectroscopy: Theory, Experiment, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Devi, P.; Sharma, C.; Kumar, P.; Kumar, M.; Bansod, B.K.S.; Nayak, M.K.; Singla, M.L. Selective electrochemical sensing for arsenite using rGO/Fe3O4 nanocomposites. J. Hazard. Mater. 2017, 322, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Peik-See, T.; Pandikumar, A.; Ngee, L.H.; Ming, H.N.; Hua, C.C. Magnetically separable reduced graphene oxide/iron oxide nanocomposite materials for environmental remediation. Catal. Sci. Technol. 2014, 4, 4396–4405. [Google Scholar] [CrossRef]
- Gao, C.; Yu, X.Y.; Xiong, S.Q.; Liu, J.H.; Huang, X.J. Electrochemical detection of arsenic (III) completely free from noble metal: Fe3O4 microspheres-room temperature ionic liquid composite showing better performance than gold. Anal. Chem. 2013, 85, 2673–2680. [Google Scholar] [CrossRef]
- Cui, H.; Yang, W.; Li, X.; Zhao, H.; Yuan, Z. An electrochemical sensor based on a magnetic Fe3O4 nanoparticles and gold nanoparticles modified electrode for sensitive determination of trace amounts of arsenic (III). Anal. Methods 2012, 4, 4176–4181. [Google Scholar] [CrossRef]
- Zhou, S.-F.; Han, X.-J.; Fan, H.-L.; Zhang, Q.-X.; Liu, Y.-Q. Electrochemical detection of As (III) through mesoporous MnFe2O4 nanocrystal clusters by square wave stripping voltammetry. Electrochim. Acta 2015, 174, 1160–1166. [Google Scholar] [CrossRef]
- Wang, L.; Xu, W.H.; Yang, R.; Zhou, T.; Hou, D.; Zheng, X.; Liu, J.H.; Huang, X.J. Electrochemical and density functional theory investigation on high selectivity and sensitivity of exfoliated nano-zirconium phosphate toward lead (II). Anal. Chem. 2013, 85, 3984–3990. [Google Scholar] [CrossRef]
- Sanllorente-Méndez, S.; Domínguez-Renedo, O.; Arcos-Martínez, M.J. Determination of Arsenic (III) Using Platinum Nanoparticle-Modified Screen-Printed Carbon-Based Electrodes. Electroanalysis 2009, 21, 635–639. [Google Scholar] [CrossRef]
- Hossain, M.M.; Islam, M.M.; Ferdousi, S.; Okajima, T.; Ohsaka, T. Anodic Stripping Voltammetric Detection of Arsenic (III) at Gold Nanoparticle-Modified Glassy Carbon Electrodes Prepared by Electrodeposition in the Presence of Various Additives. Electroanalysis 2008, 20, 2435–2441. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, Y.; Jin, H.; Zhuang, J.; Zhang, W.; Wang, S.; Wang, J. Synthesis of Au-decorated tripod-shaped Te hybrids for applications in the ultrasensitive detection of arsenic. ACS Appl. Mater. Interfaces 2013, 5, 5733–5740. [Google Scholar] [CrossRef]
- Wu, S.; Zhao, Q.; Zhou, L.; Zhang, Z. Stripping Analysis of Trace Arsenic Based on the MnOx/AuNPs Composite Film Modified Electrode in Alkaline Media. Electroanalysis 2014, 26, 1840–1849. [Google Scholar] [CrossRef]
- Li, S.S.; Zhou, W.Y.; Li, Y.X.; Jiang, M.; Guo, Z.; Liu, J.H.; Huang, X.J. Noble-Metal-Free Co0.6Fe2.4O4 Nanocubes Self-Assembly Monolayer for Highly Sensitive Electrochemical Detection of As (III) Based on Surface Defects. Anal. Chem. 2018, 90, 1263–1272. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Method | Sensitivity (μA/ppb) | Linear Range (ppb) | LOD (ppb) | References |
|---|---|---|---|---|---|
| MnOX-Au | LSASV a | 0.193 | 0.5–80 | 0.057 | [42] |
| Au-C | SWV | 0.133 | 0.375 | [26] | |
| Gr-Pt | SWASV b | 0.75–7.49 | 0.08 | [16] | |
| Pt | CV | 12–97 | 5.68 | [39] | |
| Au | ASV c | 0–87 | 1.80 | [40] | |
| RTIL-Fe3O4 | SWASV | 4.91 | 1–10 | 8 × 10 −4 | [35] |
| Co0.6Fe2.4O4 Nanocubes | SWASV | 2.12 | 1–20 | 0.093 | [43] |
| Fe3O4–rGO | SWASV | 0.281 | 0.12 | [33] | |
| Fe3O4–rGO | DPASV d | 0.142 | 2–300 | 0.10 | [25] |
| Fe3O4–rGO | SWV | 2.15 | 1–20 | 1.19 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, H.; Lu, W.; Liu, X.; Meng, F.; Zhu, J. A High-Response Electrochemical As(III) Sensor Using Fe3O4–rGO Nanocomposite Materials. Chemosensors 2021, 9, 150. https://doi.org/10.3390/chemosensors9060150
Hu H, Lu W, Liu X, Meng F, Zhu J. A High-Response Electrochemical As(III) Sensor Using Fe3O4–rGO Nanocomposite Materials. Chemosensors. 2021; 9(6):150. https://doi.org/10.3390/chemosensors9060150
Chicago/Turabian StyleHu, Haibing, Wenjie Lu, Xingnan Liu, Fancheng Meng, and Jianxiong Zhu. 2021. "A High-Response Electrochemical As(III) Sensor Using Fe3O4–rGO Nanocomposite Materials" Chemosensors 9, no. 6: 150. https://doi.org/10.3390/chemosensors9060150
APA StyleHu, H., Lu, W., Liu, X., Meng, F., & Zhu, J. (2021). A High-Response Electrochemical As(III) Sensor Using Fe3O4–rGO Nanocomposite Materials. Chemosensors, 9(6), 150. https://doi.org/10.3390/chemosensors9060150