
Pyrolyzed Photoresist Carbon Electrodes for Trace Electroanalysis of Nickel(II)


Abstract
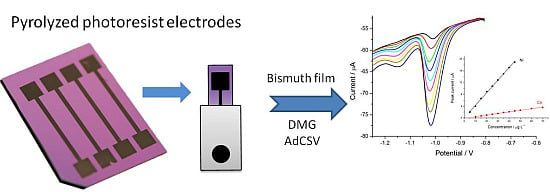
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Reagents and Materials
2.2. Apparatus
2.3. Procedure
2.3.1. PPCE Preparation
2.3.2. Analytical Procedure
3. Results and Discussion
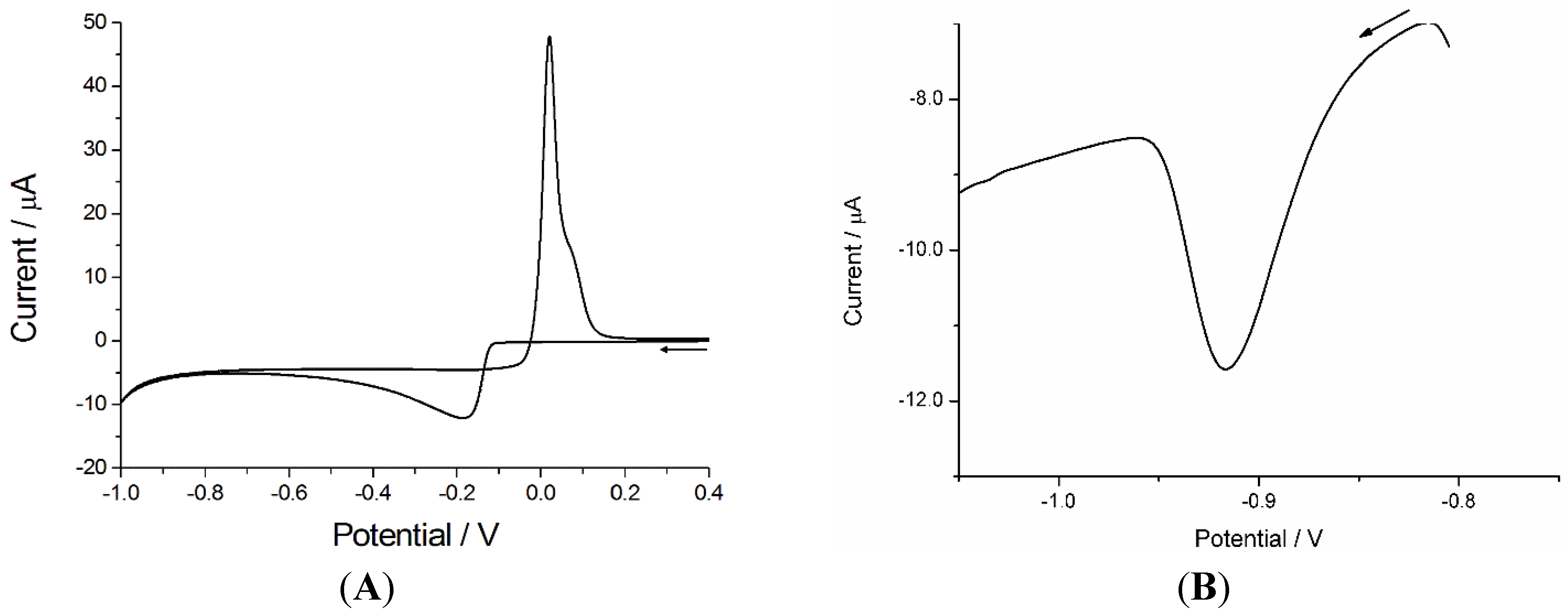
3.1. AdCSV of Ni(II) at Bi Modified PPCE
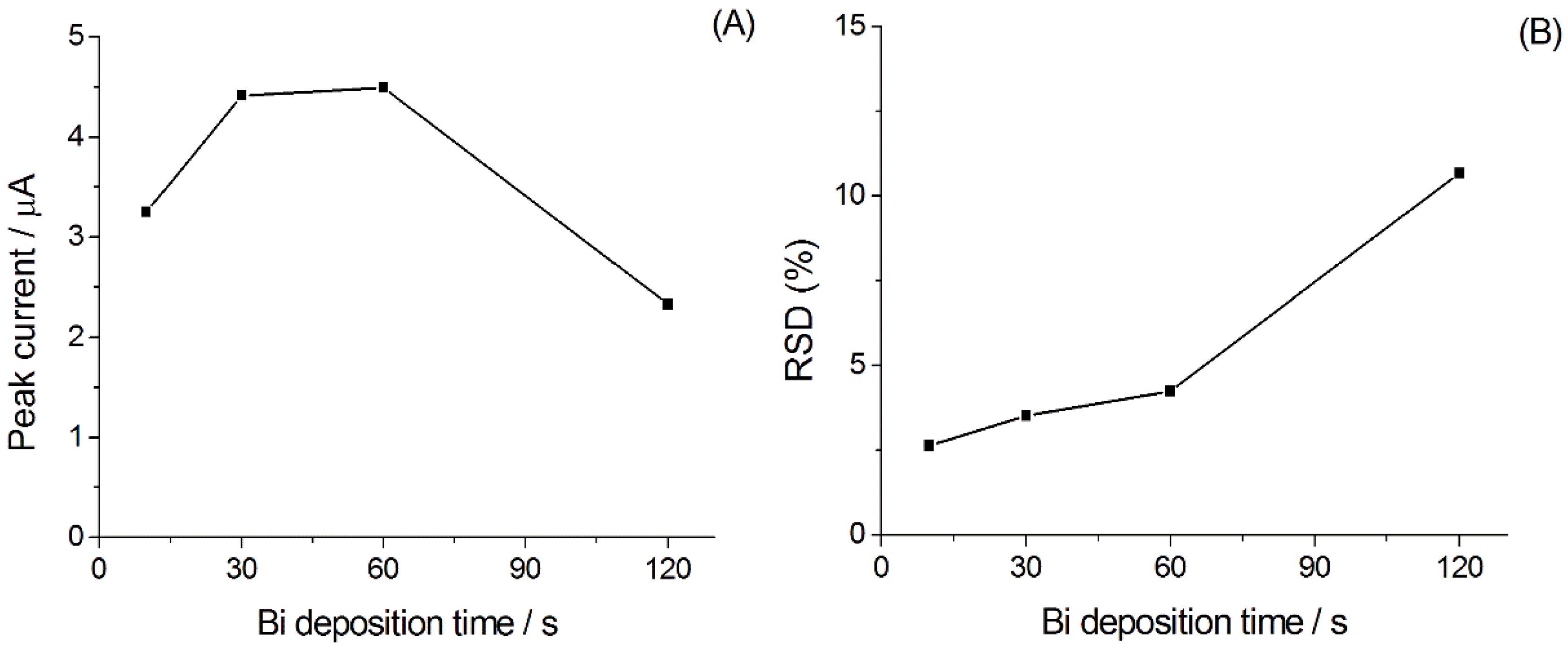
3.1.1. Optimization of the Experimental Parameters for Ni(II) Analysis
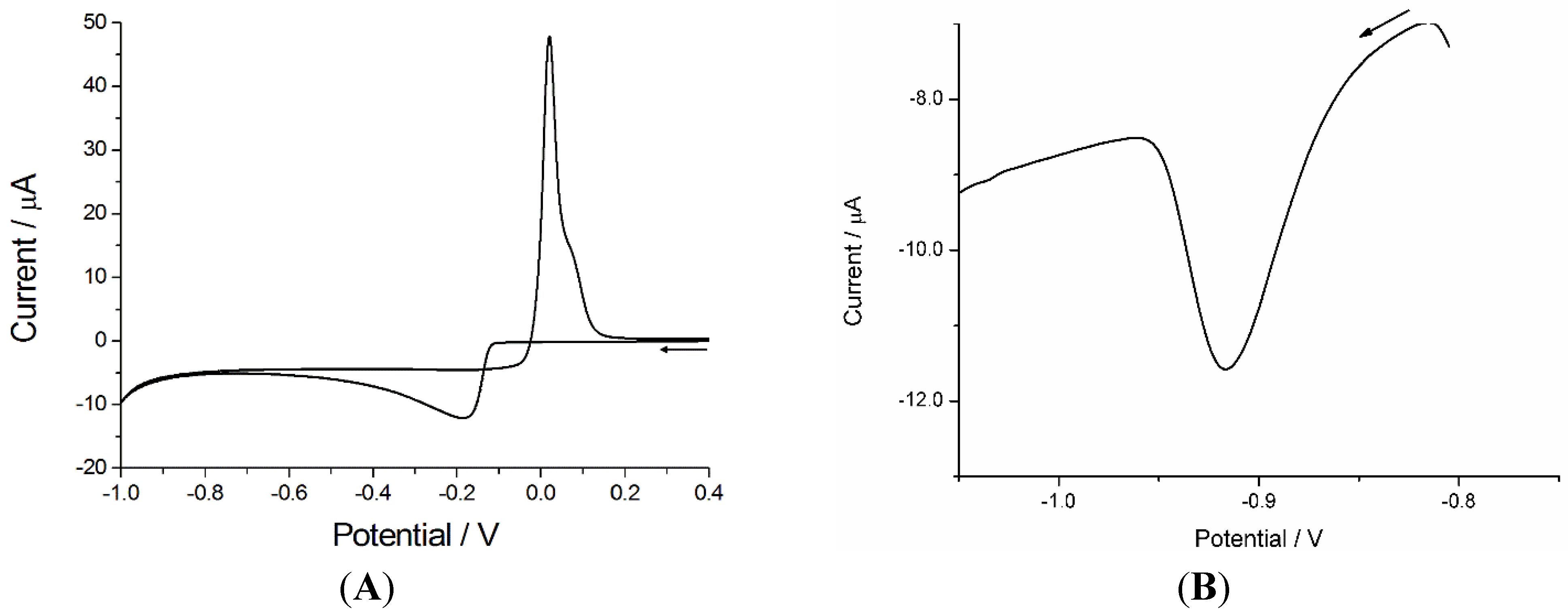
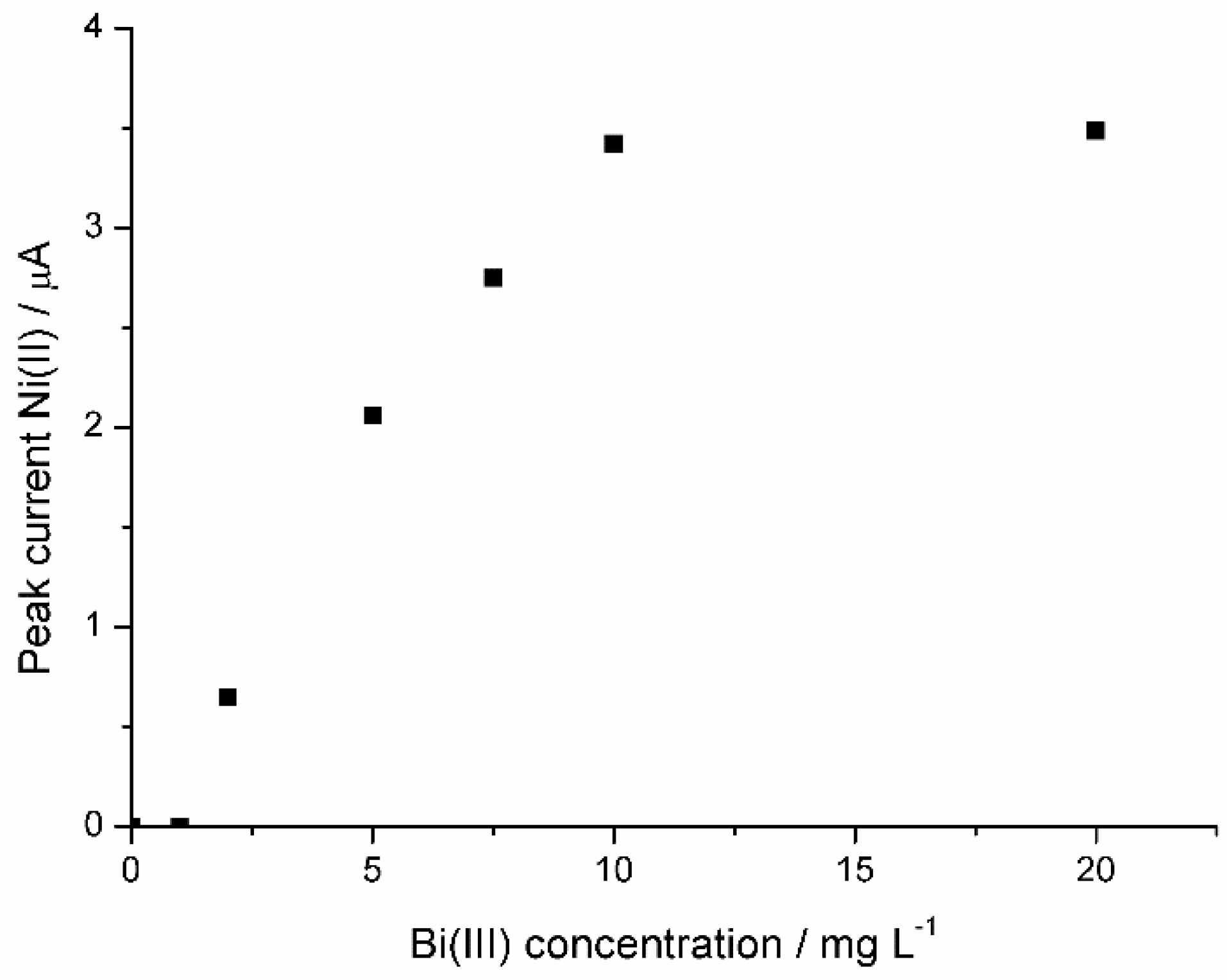
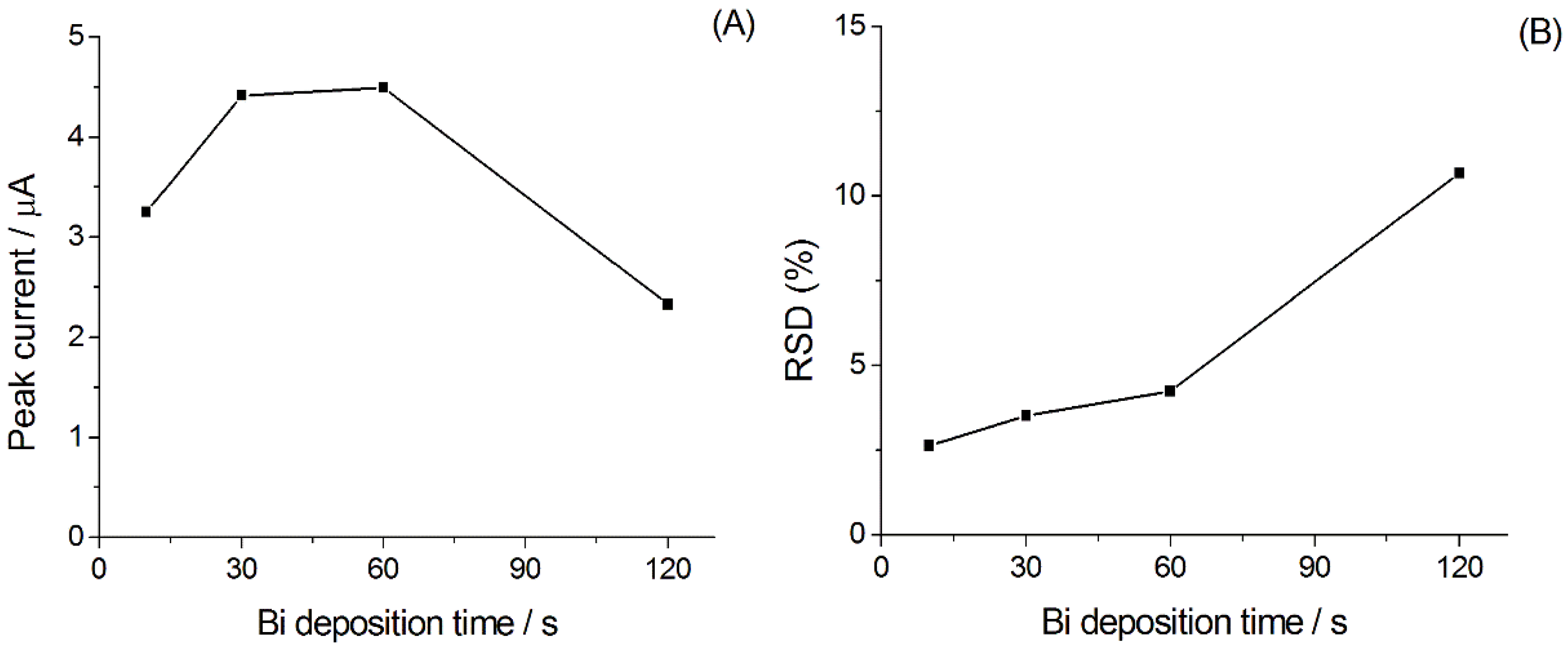
3.1.2. Calibration Plot
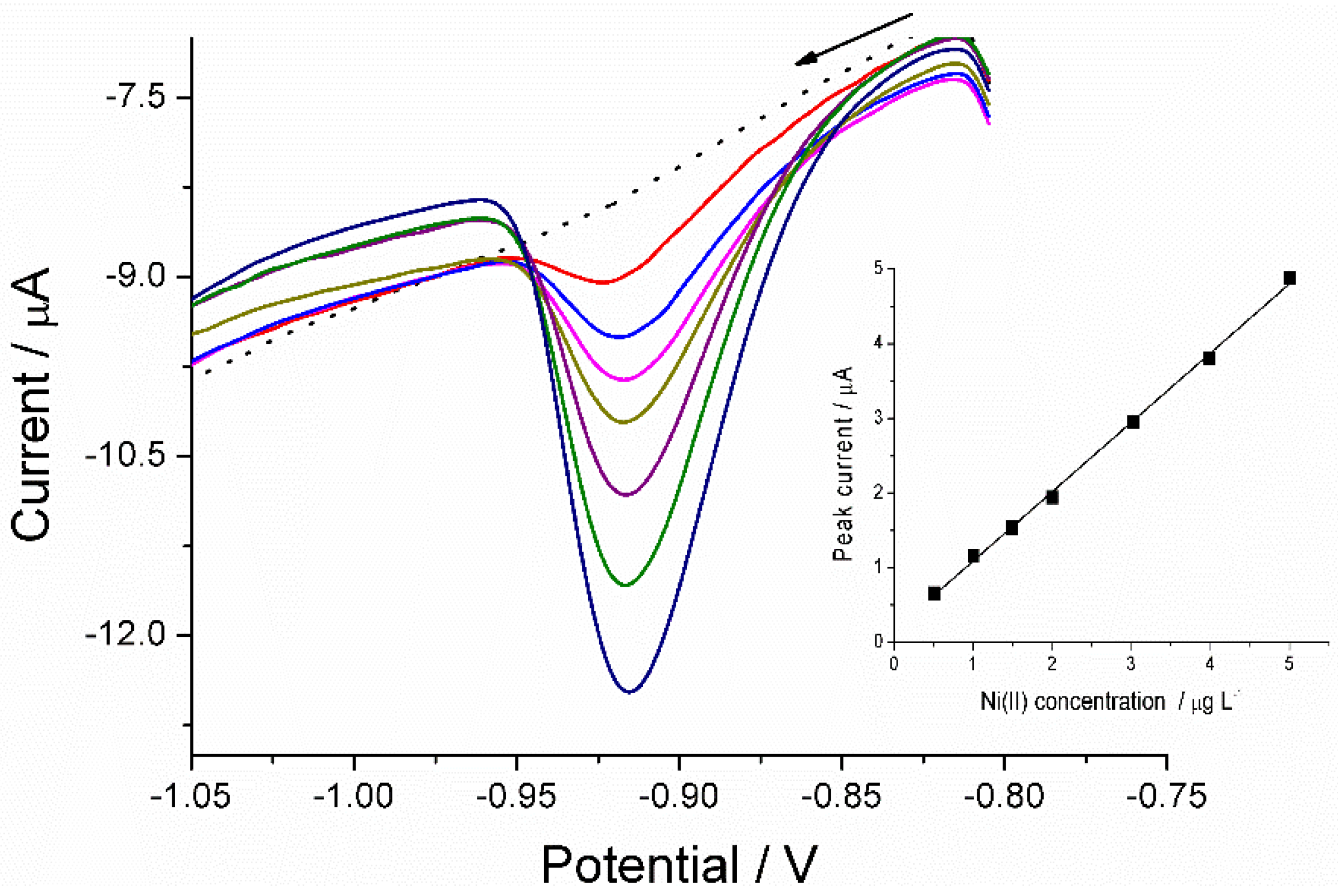
3.1.3. Adsorptive Stripping of Ni(II) in the presence of Co(II)
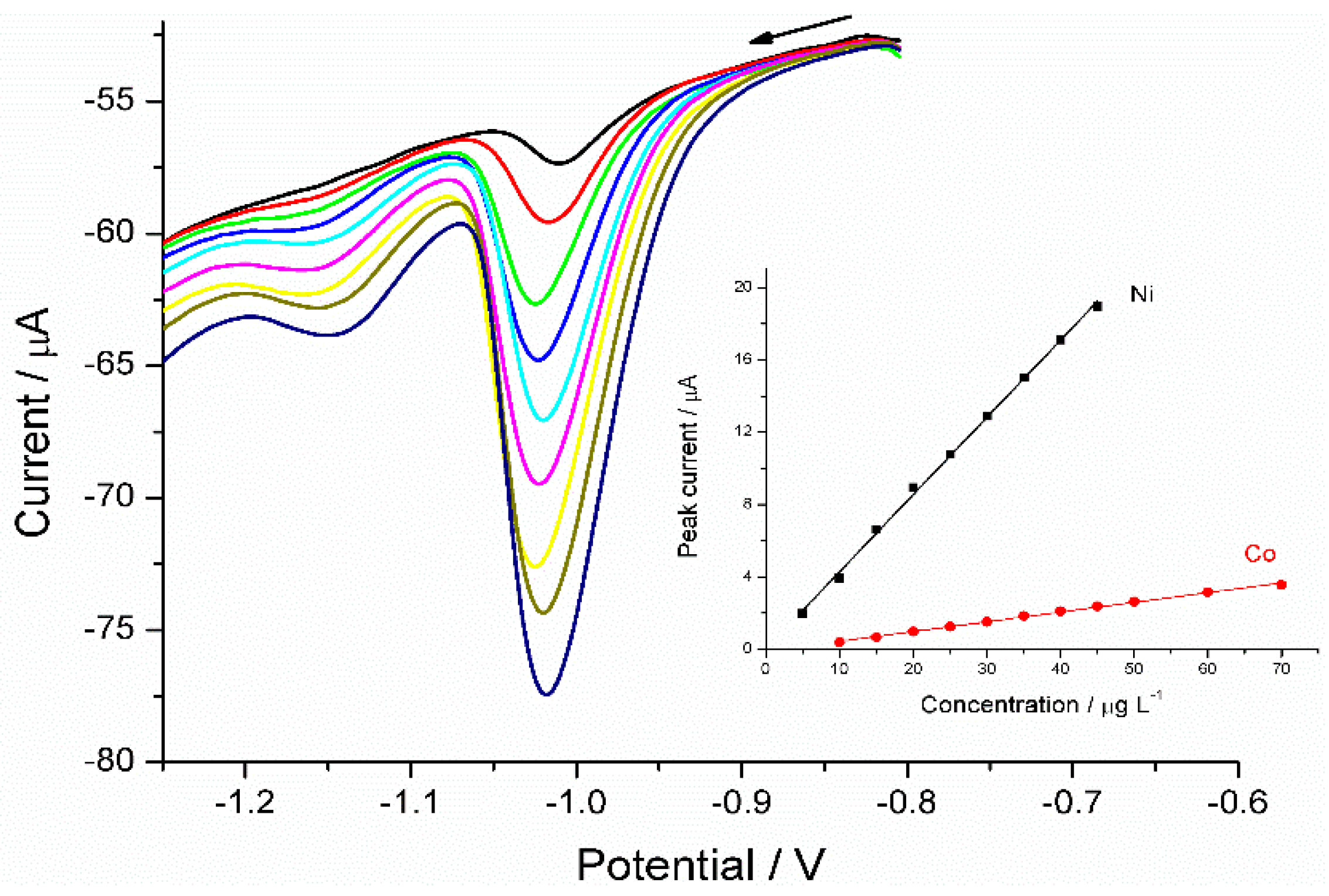
3.1.4. Analysis of Ni(II) in Certified Reference Material (NIST-1640a)

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kinoshita, K. Carbon Electrochemical and Physicochemical Properties; John Wiley & Sons: New York, NY, USA, 1988. [Google Scholar]
- Taylor, R.J.; Humffray, A.A. Electrochemical studies on glassy carbon electrodes: I. Electron transfer kinetics. J. Electroanal. Chem. Interfacial Electrochem. 1973, 42, 347–354. [Google Scholar] [CrossRef]
- Florence, T.M. Anodic stripping voltammetry with a glassy carbon electrode mercury-plated in situ. J. Electroanal. Chem. Interfacial Electrochem. 1970, 27, 273–281. [Google Scholar] [CrossRef]
- Sun, Y.C.; Mierzwa, J.; Yang, M.H. New method of gold-film electrode preparation for anodic stripping voltammetric determination of arsenic (III and V) in seawater. Talanta 1997, 44, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Svancara, I.; Prior, C.; Hocevar, S.B.; Wang, J. A decade of bismuth-based electrodes in electroanalysis. Electroanalysis 2010, 22, 1405–1420. [Google Scholar] [CrossRef]
- Hocevar, S.B.; Svancara, I.; Ogorevc, B.; Vytras, K. Antimony film electrode for electrochemical stripping analysis. Anal. Chem. 2007, 79, 8639–8643. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Nekrassova, O.; Hyde, M.E.; Compton, R.G. Anodic stripping voltammetry of arsenic(III) using gold nanoparticle-modified electrodes. Anal. Chem. 2004, 76, 5924–5929. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Compton, R.G. Direct electrodeposition of gold nanoparticles onto indium tin oxide film coated glass: Application to the detection of arsenic(III). Anal. Sci. 2006, 22, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Ugo, P.; Moretto, L.M.; Mazzocchin, G.A. Voltammetric determination of trace mercury in chloride media at glassy carbon electrodes modified with polycationic ionomers. Anal. Chim. Acta 1995, 305, 74–82. [Google Scholar]
- Ugo, P.; Moretto, L.M. Ion-exchange voltammetry at polymer-coated electrodes: Principles and analytical prospects. Electroanalysis 1995, 7, 1105–1113. [Google Scholar] [CrossRef]
- Zachek, M.K.; Takmakov, P.; Moody, B.; Wightman, R.M.; McCarty, G.S. Simultaneous decoupled detection of dopamine and oxygen using pyrolyzed carbon microarrays and fast-scan cyclic voltammetry. Anal. Chem. 2009, 81, 6258–6265. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Song, X.; Kinoshita, K.; Madou, M.; White, R. Electrochemical Studies of Carbon Films from Pyrolyzed Photoresist. J. Electrochem. Soc. 1998, 145, 2314–2319. [Google Scholar] [CrossRef]
- Ranganathan, S.; McCreery, R.; Majji, S.M.; Madou, M. Photoresist derived carbon for microelectromechanical systems and electrochemical applications. J. Electrochem. Soc. 2000, 147, 277–282. [Google Scholar] [CrossRef]
- Mardegan, A.; Kamath, R.; Sharma, S.; Scopece, P.; Ugo, P.; Madou, M. Optimization of carbon electrodes derived from epoxy-based photoresist. J. Electrochem. Soc. 2013, 160, B132–B137. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Hocevar, S.B.; Farias, P.A.M.; Ogorevc, B. Bismuth-coated carbon electrodes for anodic stripping voltammetry. Anal. Chem. 2000, 72, 3218–3222. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, Ø.; Schrøder, K.H. An Oscillating and renewing silver electrode for cadmium and lead detection in differential pulse stripping voltammetry. Electroanalysis 2001, 13, 687–692. [Google Scholar]
- Nolan, M.A.; Kounaves, S.P. Microfabricated array of iridium microdisks as a substrate for direct determination of Cu2+ or Hg2+ using square-wave anodic stripping voltammetry. Anal. Chem. 1999, 71, 3567–3573. [Google Scholar] [CrossRef]
- Mikkelsen, Ø.; Skogvold, S.M.; Schrøder, K.H.; Gjerde, M.I.; Aarhaug, T.A. Evaluation of solid electrodes for use in voltammetric monitoring of heavy metals in samples from metallurgical nickel industry. Anal. Bioanal. Chem. 2003, 377, 322–326. [Google Scholar]
- Švancara, I.; Matoušek, M.; Sikora, E.; Schachl, K.; Kalcher, K.; Vytřas, K. Carbon paste electrodes plated with a gold film for the voltammetric determination of mercury(II). Electroanalysis 1997, 9, 827–833. [Google Scholar] [CrossRef]
- Charalambous, A.; Economou, A. A study on the utility of bismuth-film electrodes for the determination of In(III) in the presence of Pb(II) and Cd(II) by square wave anodic stripping voltammetry. Anal. Chim. Acta 2005, 547, 53–58. [Google Scholar] [CrossRef]
- Švancara, I.; Baldrianová, L.; Tesařová, E.; Hočevar, S.B.; Elsuccary, S.A.A.; Economou, A.; Sotiropoulos, S.; Ogorevc, B.; Vytřas, K. Recent advances in anodic stripping voltammetry with bismuth-modified carbon paste electrodes. Electroanalysis 2006, 18, 177–185. [Google Scholar] [CrossRef]
- Mardegan, A.; Dal Borgo, S.; Scopece, P.; Moretto, L.M.; Hočevar, S.B.; Ugo, P. Bismuth modified gold nanoelectrode ensemble for stripping voltammetric determination of lead. Electrochem. Commun. 2012, 24, 28–31. [Google Scholar] [CrossRef]
- Bučková, M.; Gründler, P.; Flechsig, G.U. Adsorptive stripping voltammetric detection of daunomycin at a bismuth bulk electrode. Electroanalysis 2005, 17, 440–444. [Google Scholar] [CrossRef]
- Hutton, E.A.; Hočevar, S.B.; Ogorevc, B.; Smyth, M.R. Bismuth film electrode for simultaneous adsorptive stripping analysis of trace cobalt and nickel using constant current chronopotentiometric and voltammetric protocol. Electrochem. Commun. 2003, 5, 765–769. [Google Scholar] [CrossRef]
- Paneli, M.G.; Voulgaropoulos, A. Applications of adsorptive stripping voltammetry in the determination of trace and ultratrace metals. Electroanalysis 1993, 5, 355–373. [Google Scholar] [CrossRef]
- Brett, C.M.A.; Oliveira Brett, A.M.C.F.; Pereira, J.L.C. Adsorptive stripping voltammetry of cobalt and nickel in flow systems at wall-jet electrodes. Electroanalysis 1991, 3, 683–689. [Google Scholar] [CrossRef]
- Wang, J. Analytical Electrochemistry; Wiley-VCH: Verlag, Germany, 2006. [Google Scholar]
- Hutton, E.A.; Ogorevc, B.; Hočevar, S.B.; Smyth, M.R. Bismuth film microelectrode for direct voltammetric measurement of trace cobalt and nickel in some simulated and real body fluid samples. Anal. Chim. Acta 2006, 557, 57–63. [Google Scholar] [CrossRef]
- Korolczuk, M.; Rutyna, I.; Tyszczuk, K. Adsorptive stripping voltammetry of Nickel at an in-situ plated Bismuth film electrode. Electroanalysis 2010, 22, 1494–1498. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J. Bismuth film electrodes for adsorptive stripping voltammetry of trace nickel. Electrochem. Commun. 2000, 2, 390–393. [Google Scholar] [CrossRef]
- Ruhlig, D.; Schulte, A.; Schuhmann, W. An electrochemical robotic system for routine cathodic adsorptive stripping analysis of Ni2+ ion release from corroding NiTi shape memory alloys. Electroanalysis 2006, 18, 53–58. [Google Scholar] [CrossRef]
- Piankova, L.A.; Malakhova, N.A.; Stozhko, N.Y.; Brainina, K.Z.; Murzakaev, A.M.; Timoshenkova, O.R. Bismuth nanoparticles in adsorptive stripping voltam-metry of nickel. Electrochem. Commun. 2011, 13, 981–984. [Google Scholar] [CrossRef]
- Alves, G.M.S.; Magalhães, J.M.C.S.; Soares, H.M.V.M. Simultaneous determination of nickel and cobalt, using a solid bismuth vibrating electrode, by adsorptive cathodic stripping voltammetry. Electroanalysis 2013, 25, 1247–1255. [Google Scholar] [CrossRef]
- Mardegan, A.; Dal Borgo, S.; Scopece, P.; Moretto, L.M.; Hocevar, S.B.; Ugo, P. Simultaneous adsorptive cathodic stripping voltammetric determination of Nickel(II) and Cobalt(II) at the in-situ bismuth-modified gold electrode. Electroanalysis 2013, 25, 2471–2479. [Google Scholar] [CrossRef]
- Sopha, H.; Jovanovski, V.; Hocevar, S.B.; Ogorevc, B. In-situ plated antimony film electrode for adsorptive cathodic stripping voltammetric measurement of trace nickel. Electrochem. Commun. 2012, 20, 23–25. [Google Scholar] [CrossRef]
- SU-8 2000 permanent epoxy negative photoresist processing guidelines for:SU-8 2025, SU-8 2035, SU-8 2050 and SU-8 2075. Available online: http://www.microchem.com/pdf/SU-82000DataSheet2025thru2075Ver4.pdf (accessed on 12 May 2015).
- Korolczuk, M.; Moroziewicz, A.; Grabarczyk, M. Determination of subnanomolar concentrations of cobalt by adsorptive stripping voltammetry at a bismuth film electrode. Anal. Bioanal. Chem. 2005, 382, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Hu, Z. Electrodeposition of bismuth onto glassy carbon electrodes from nitrate solutions. J. Electroanal. Chem. 2005, 583, 46–55. [Google Scholar] [CrossRef]
- Ma, F.; Jagner, D.; Renman, L. Mechanism for the electrochemical stripping reduction of the nickel and cobalt dimethylglyoxime complexes. Anal. Chem. 1997, 69, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Rutyna, I.; Korolczuk, M. Catalytic adsorptive stripping voltammetry of cobalt in the presence of nitrite at an in situ plated bismuth film electrode. Electroanalysis 2011, 23, 637–641. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretto, L.M.; Mardegan, A.; Cettolin, M.; Scopece, P. Pyrolyzed Photoresist Carbon Electrodes for Trace Electroanalysis of Nickel(II). Chemosensors 2015, 3, 157-168. https://doi.org/10.3390/chemosensors3020157
Moretto LM, Mardegan A, Cettolin M, Scopece P. Pyrolyzed Photoresist Carbon Electrodes for Trace Electroanalysis of Nickel(II). Chemosensors. 2015; 3(2):157-168. https://doi.org/10.3390/chemosensors3020157
Chicago/Turabian StyleMoretto, Ligia Maria, Andrea Mardegan, Mattia Cettolin, and Paolo Scopece. 2015. "Pyrolyzed Photoresist Carbon Electrodes for Trace Electroanalysis of Nickel(II)" Chemosensors 3, no. 2: 157-168. https://doi.org/10.3390/chemosensors3020157
APA StyleMoretto, L. M., Mardegan, A., Cettolin, M., & Scopece, P. (2015). Pyrolyzed Photoresist Carbon Electrodes for Trace Electroanalysis of Nickel(II). Chemosensors, 3(2), 157-168. https://doi.org/10.3390/chemosensors3020157