1. Introduction
Glioblastoma is an aggressive and fatal form of brain cancer that primarily affects adults [
1]. Despite standard treatment protocols, which typically involve surgery followed by radiation and chemotherapy, the prognosis remains poor, with overall survival averaging only a few months [
2]. Gold standard techniques, such as tissue biopsies and imaging techniques, are routinely employed to characterize tumors and predict treatment responses [
3,
4,
5], but tumor resection and biopsy carry significant risks, including the potential for neurological deficits in the patient [
6,
7]. In this context, the potential of liquid biopsies in GBM management is gaining increasing interest [
8,
9], and further analysis of these cells may greatly aid in both research and clinical applications.
Liquid biopsy involves the analysis of cells from biofluids such as blood, urine, and saliva [
10,
11]. This minimally invasive technique aims to provide real-time insights into tumor dynamics, enabling disease progression diagnosis and monitoring. Tumor-derived material can be shed into the bloodstream, including circulating tumor DNA (ctDNA) and circulating tumor cells. CTCs, which detach from the primary tumor and enter circulation, play a role in metastasis and have been shown to serve as biomarkers for predicting disease outcomes and monitoring treatment responses [
12,
13]. Recent studies have demonstrated that GBM can release tumor-related material into circulation [
14,
15], and CTCs have been detected in the blood of GBM patients [
16,
17,
18]. The ability to detect these tumor cells in peripheral blood offers a non-invasive alternative to traditional biopsy, facilitating the monitoring of tumor characteristics and treatment response without more invasive procedures.
A variety of techniques have been developed for the isolation and detection of circulating tumor cells, ranging from immunomagnetic bead-based methods [
19,
20,
21], such as the CellSearch platform by Veridex (10), to size-based filtration systems [
22] and microfluidic devices [
23,
24,
25,
26]. Combining size-based filtration and immunoaffinity for circulating tumor cell isolation has been extensively explored as a synergistic approach to improve detection specificity and efficiency. Juncker and co-workers demonstrated the potential of this dual strategy by utilizing a membrane filter functionalized with specific antibodies targeting tumor cell surface markers. This methodology leverages the physical property of tumor cells larger than most blood cells, allowing their separation through size exclusion. At the same time, the antibody-functionalized surface enhances capture efficiency and specificity by selectively binding to antigens expressed on tumor cells [
25,
27]. Although these methods improve the likelihood of interactions between CTCs and antibodies, they fall short of addressing the cloaking effect. This phenomenon occurs when a CTC is loosely enveloped by platelets and other blood cells, forming a protective shield that hinders direct contact with therapeutic agents or detection tools. The cloaking effect reduces the efficacy of antibody binding and complicates efforts to isolate and characterize CTCs accurately. Overcoming this challenge remains critical for advancing diagnostic precision and therapeutic interventions targeting CTCs. The integration of size filtration with immunoaffinity holds significant promise for advancing liquid biopsy technologies and improving early cancer detection and monitoring. Dr. Fan’s group recently introduced an innovative lateral filter array (LFA) microfluidic device that combines size-based separation with immunoaffinity-based CTC isolation and was applied to patients with metastatic pancreatic cancer. This dual-function approach addresses the limitations of existing platforms that rely solely on a single isolation mechanism [
27]. LFA incorporates lateral filters ranging from 10–30 µm in size, specifically designed to filter CTCs. This approach combines the physical property of size with the biological property of immunoaffinity for more efficient isolation. The study explored integrating size-based microfiltration with immunoaffinity within a microfluidic device to improve CTC capture efficiency. The device features four serpentine main channels, each equipped with lateral filter arrays that generate a two-dimensional flow. This design demonstrated significantly higher CTC capture efficiency compared to filters lacking antibody integration. In this pilot study, we further explored using the LFA device, which contains 12 to 24 µm microfilters [
28], to enhance CTC capture efficiency in patients diagnosed with GBM before surgery. In the LFA device, the microfilters function more as a “gate” than a traditional filter, selectively blocking more giant cells while allowing smaller CTCs to interact with the antibody-coated surfaces. The device’s design was optimized to maximize the interaction between the CTCs and the filters, ensuring high capture efficiency. This study is the first to demonstrate successful CTC enrichment from the peripheral blood of GBM patients using the LFA microfluidic chip, offering a promising tool for non-invasive cancer monitoring and diagnostics.
2. Experimental Section
2.1. Reagents and Materials
The reagents used in this study were obtained from Sigma-Aldrich (St. Louis, MO, USA) and utilized without further modification, including Sylgard 184, ethanol, and Dulbecco’s phosphate-buffered saline (DPBS) containing calcium chloride and magnesium chloride, Bovine Serum Albumin (BSA), EDTA, DPBS, Avidin, Triton X-100, paraformaldehyde, anti-CD45-PE, anti-cytokeratin-FITC, and DAPI were used as received without further purification. Anti-EpCAM (Anti-Human CD326) was purchased from eBioscience, San Diego, CA, USA.
2.2. Fabrication of the LFA Device
The LFA device was fabricated using a previously established method [
28]. The channel pattern was initially designed in AutoCAD, and the device consisted of a 1-in × 3 in. glass slide bonded to a poly(dimethylsiloxane) (PDMS) layer featuring four microchannels. A PDMS substrate was created through soft lithography using a silicon master mold. A thoroughly mixed PDMS solution, prepared at a 10:1 base-to-curing agent ratio, was poured onto the silicon master. To eliminate air bubbles, the PDMS mixture was degassed in a vacuum chamber before being cured in an oven at 65 °C for at least 4 h. After curing, the PDMS formed a flexible and transparent substrate, carefully peeled from the silicon master, trimmed to the appropriate size, and perforated at designated inlet and outlet locations. Finally, the PDMS substrate was bonded to the glass slide using a 5-min UV ozone treatment to ensure a robust and leak-proof seal. Geometry layout of the LFA device. The LFA device features four serpentine main channels, each integrated with an array of lateral filters (
Scheme 1A). These filters are organized into 11 distinct zones, categorized by filter size. Each zone contains 10 columns of lateral filters of uniform size. The largest filter zone, with a filter size of 23.8 µm, is positioned near the device’s inlet, while the smallest filter zone, measuring 12.3 µm, is located closer to the outlet. The width of each serpentine channel is 300 µm, with individual filter lengths (L1) of 50 µm. The filter widths (W1) vary from 12.3 µm to 23.8 µm (
Scheme 1B). The spacing between lateral filters (W2) can be either 50 µm or 100 µm, determined by the number of filters in the column.
2.3. Functionalization of the LFA Device
The microchannels were cleaned in alcohol and filled with a 20 µg/mL avidin solution prepared in DPBS containing calcium chloride and magnesium chloride to establish a specific biorecognition layer. The devices were incubated for 30 min, allowing the avidin molecules to adsorb onto the PDMS surfaces. Following incubation, the channels were rinsed with 200 µL of DPBS to remove unbound avidin, ensuring that only firmly attached molecules remained. For the final functionalization step, anti-EpCAM antibodies were immobilized using biotin-avidin interactions. Then, 70 µL of biotinylated anti-EpCAM antibody solution (10 µg/mL) was added and incubated for 30 min, followed by 200 µL of DPBS to remove unbound reaction. This step ensured the biotinylated antibodies specifically bound to the avidin molecules through strong biotin-avidin interactions, creating a stable and oriented antibody layer crucial for effective antigen capture. Before the experiment, the LFA device was passivated to reduce nonspecific interactions. This was achieved by filling the channels with a blocking buffer containing 1% BSA in DPBS. The BSA treatment blocked residual nonspecific binding sites, minimizing background noise and preventing the unwanted capture of normal blood cells.
2.4. Cell Culture
The pancreas cancer cell lines Hs578t and CCRF-CEM were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cell lines were expanded under standard cell culture conditions and used when they reached 85% confluence. The cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) from ATCC, supplemented with 10% fetal bovine serum (FBS; GIBCO, Thermo Fisher, Waltham, MA, USA) and 100 units/mL penicillin-streptomycin (Cellgro, Manassas, VA, USA). The incubation conditions were set at 37 °C in a humidified atmosphere containing 5% CO2. The CCRF-CEM was cultured in RPMI 1640 medium (ATCC) with 10% FBS and 100 units/mL penicillin-streptomycin.
For Hs578t, the culture medium was carefully removed from the flask, and the cells were rinsed with DPBS to eliminate residual impurities. A 2 mL volume of 0.25% trypsin-EDTA (GIBCO, Fisher Scientific) was added, and the flask was incubated for 10 min to detach the cells. A growth medium (6 mL) was then added to neutralize the trypsin, and the cell suspension was transferred to a tube. The cells were centrifuged and washed twice with DPBS to ensure the removal of any remaining impurities. Finally, the cells were resuspended in 1 mL of DPBS for subsequent use. For the suspension cell line (CCRF-CEM), cells were withdrawn from the culture flask and washed twice with DPBS. The cells were then resuspended in 1 mL of DPBS. Both cell types were stained using Vybrant dyes (Thermo Fisher, Waltham, MA, USA) following the manufacturer’s instructions. The stained cells were washed with DPBS and used to spike into DPBS buffer or blood samples for experiments.
2.5. Tumor Cell Capture in Devices
The optimization step was carried out with Hs578t cells as a model. Then, 500–5000 cells were spiked into 1 mL of DPBS for buffer-based experiments. The spiked samples were infused into the LFA device at flow rates ranging from 0.5 mL/h to 1.5 µL/s. Following sample infusion, 250 µL of DPBS was used to wash away unbound cells or impurities from the device. Tumor cells captured in the device were visualized and recorded using an Olympus IX71 fluorescence microscope (Olympus America, Center Valley, PA, USA) equipped with a Hamamatsu C4742-80-12AG scientific-grade CCD camera. For devices not functionalized with antibodies, cells were released by pumping DPBS from the outlet at a high flow rate of 10 mL/h and collected from the inlet. For antibody-functionalized devices, the captured cells were treated with one channel volume of 0.25% trypsin-EDTA for 10 min to detach them. Subsequently, DPBS was pumped from the outlet at 10 mL/h to collect the released cells.
2.6. Clinical Samples
Blood samples were obtained from glioblastoma patients, followed by the Neurosurgery Service at the Clinics Hospital of Botucatu Medical School, UNESP, Brazil. The collection and handling of specimens followed protocols approved by the Research Ethics Committee of Botucatu Medical School—UNESP (CAAE: 47991921.0.0000.5411) and were conducted by the Declaration of Helsinki. The study included three patients: one female (43 years old) and two males (54 and 48 years old, respectively); cases were IDH wild-type GBM. All samples were processed within four hours of collection. Before the arrival of the clinical samples, two LFA devices were functionalized with anti-EpCAM and passivated using a 1% BSA solution, as previously described. Before the arrival of clinical samples, two LFA devices were functionalized with anti-EpCAM and passivated using a 1% BSA solution, as described earlier. The collected blood samples were diluted 1:1 with DPBS and infused into the antibody-functionalized devices at a 1.5 µL/s flow rate. A total volume of 8 mL of diluted blood was processed using two devices in parallel (4 mL per device). After the sample infusion, each device was washed with five channel volumes of DPBS to remove impurities. Cell fixation was carried out by introducing one channel volume of 4% paraformaldehyde (PFA) solution and incubating for 10 min. The devices were then washed, and cell membrane permeabilization was achieved by introducing one channel volume of 0.2% Triton X-100 solution and incubating for 10 min.
For immunostaining, a mixture containing 10 μg/mL anti-CD45-PE, 10 μg/mL anti-cytokeratin-FITC, and 500 nM DAPI was introduced into each device and incubated for 30 min. The devices were subsequently washed and mounted on the fluorescence microscope stage for circulating tumor cell (CTC) enumeration. CTCs were identified as cells that were positive for DAPI (nuclear stain), negative for CD45 (leukocyte marker), and positive for cytokeratin (epithelial marker). Other cell types, such as white blood cells and any cells with atypical staining patterns, were excluded from the analysis.
3. Results and Discussion
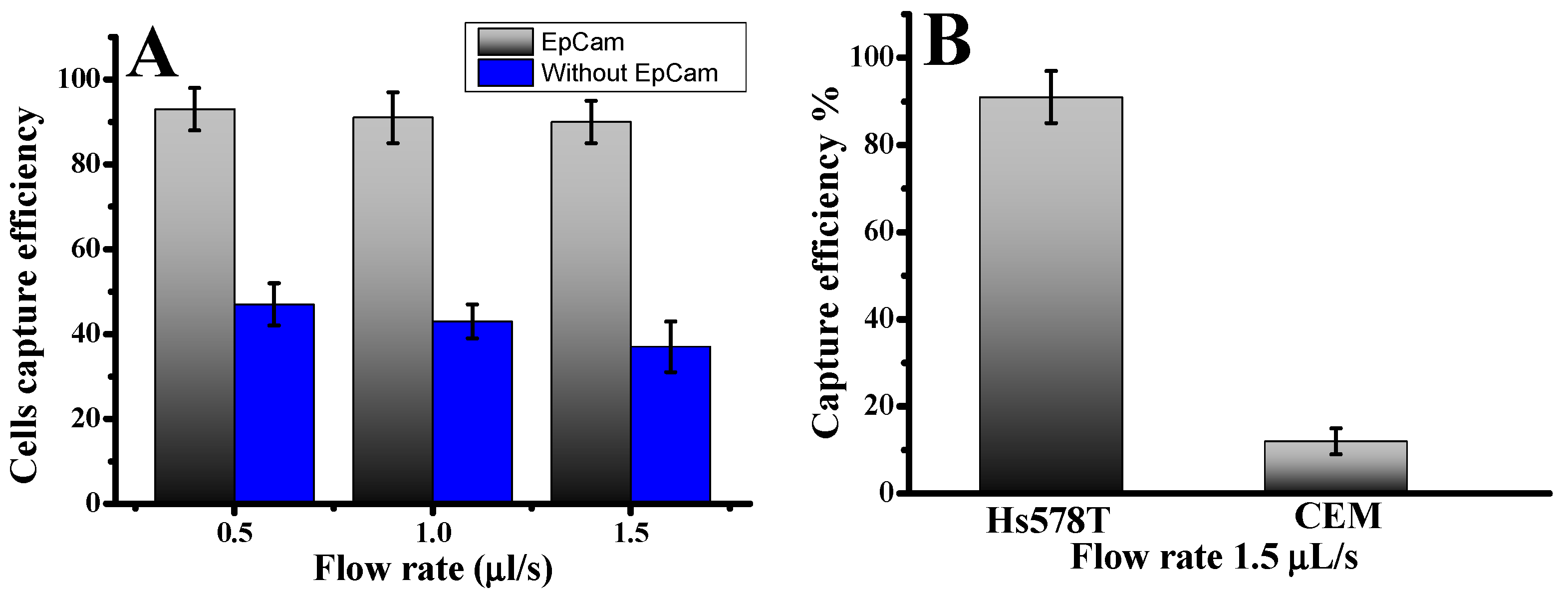
Approximately 1000 fluorescence-labeled Hs578t cells were infused into the device to investigate CTC capture using our microfluidic laminar flow device. The diameters of Hs578t cells were 13.0 ± 4.0 µm. The results illustrate the cell capture ratios with an anti-EpCAM-coated LFA device and without under varying flow rates. The cell capture patterns in the anti-EpCAM-coated LFA device were compared to those in an LFA device without anti-EpCAM coating. As illustrated in
Figure 1A, cell capture peaks at varying infused flow rates were consistently located between the 15.7 µm and 12.3 µm filter zones, where the filter pore sizes were smaller than the cell diameters [
28]. Without anti-EpCAM immobilization, the LFA device exhibited low circulating tumor cell capture efficiency (
Figure 1), capturing only 38–43% of both cell types even at a low flow rate of 0.5 µL/s. In contrast, functionalizing the chip with anti-EpCAM dramatically improved capture efficiency. The capture efficiency rose to 93.0 ± 3.0% at 0.5 µL/s. This significant enhancement underscores the predominance of affinity-based mechanisms in cell capture when using an anti-EpCAM functionalized LFA device. Such results demonstrate the critical role of EpCAM-mediated binding in overcoming limitations of size-exclusion-based capture. Previous studies have also highlighted the superior performance of EpCAM-functionalized platforms in isolating CTCs with high specificity and efficiency, particularly at lower flow rates, where binding kinetics are more favorable [
28]. The CTC capture efficiency in the microfluidic system was determined using the formula: Cf/Cs × 100%, where Cf represents the number of cancer cells identified under a fluorescence microscope, and Cs is the total number of cancer cells added to the DPBS buffer. These results confirm the effectiveness of combining size-based filtration with antibody-mediated affinity capture, providing a more effective and accurate strategy for CTC isolation.
To evaluate cell capture efficiency across different cell types and sizes, we investigated a mixture of cancer cell lines: the target Hs578t cells (EpCAM+) and control CCRF-CEM cells (EpCAM−). As shown in
Figure 1B, the capture efficiency for Hs578t cells reached 91.0 ± 4.5% at a flow rate of 1.5 µL/s. In contrast, most CCRF-CEM cells (13.0 ± 2 µm in diameter) were removed during the washing process, with a capture efficiency of only 10.0 ± 4%. This disparity can be attributed to the absence of EpCAM expressions in CCRF-CEM cells and their smaller size, which reduces their likelihood of being captured. These findings highlight the effectiveness of our device, which combines nanoparticles and anti-EpCAM functionalization, in selectively isolating circulating tumor cells even at low concentrations. The release of captured cells was accomplished using trypsinization. Release efficiency was determined as the ratio of released cells to captured cells. Overall, 78.0 ± 4% of the captured cells remained viable after the capture and release process, demonstrating their suitability for subsequent cellular analysis. This platform offers a reliable method for isolating EpCAM-positive cells with high specificity by minimizing nonspecific surface interactions and enhancing binding efficiency.
The technical workflow for CTC enrichment is illustrated in
Figure 2. Previous studies have demonstrated that adding bovine serum albumin and EDTA to the priming buffer significantly enhances CTC recovery by preventing cell adhesion. To evaluate the effect of buffer composition on cell recovery using fixed samples, we spiked 5.000 Hs578t cancer cells into 2 mL of healthy donor blood. We performed enrichment using the standard DPBS, and DPBS was supplemented with 1% BSA and 1 mM EDTA for cell separation. Results revealed significantly higher recovery rates for Hs578t when using DPBS supplemented with 1% BSA and 1 mM EDTA compared to DPBS alone, with mean recoveries of 84.5 ± 4% and 70.0 ± 3%, respectively. Furthermore, adding only BSA, the capture efficiency achieved was 78.0 ± 4%. These findings suggest that including BSA and EDTA enhances the recovery efficiency of CTCs. This improvement may stem from the anti-adhesive properties of BSA, which coats surfaces and prevents nonspecific cell binding, and EDTA, which chelates divalent cations critical for cell-surface interactions. This optimized buffer composition ensures higher yield and integrity of isolated cells, facilitating more accurate downstream analysis and characterization of CTCs.
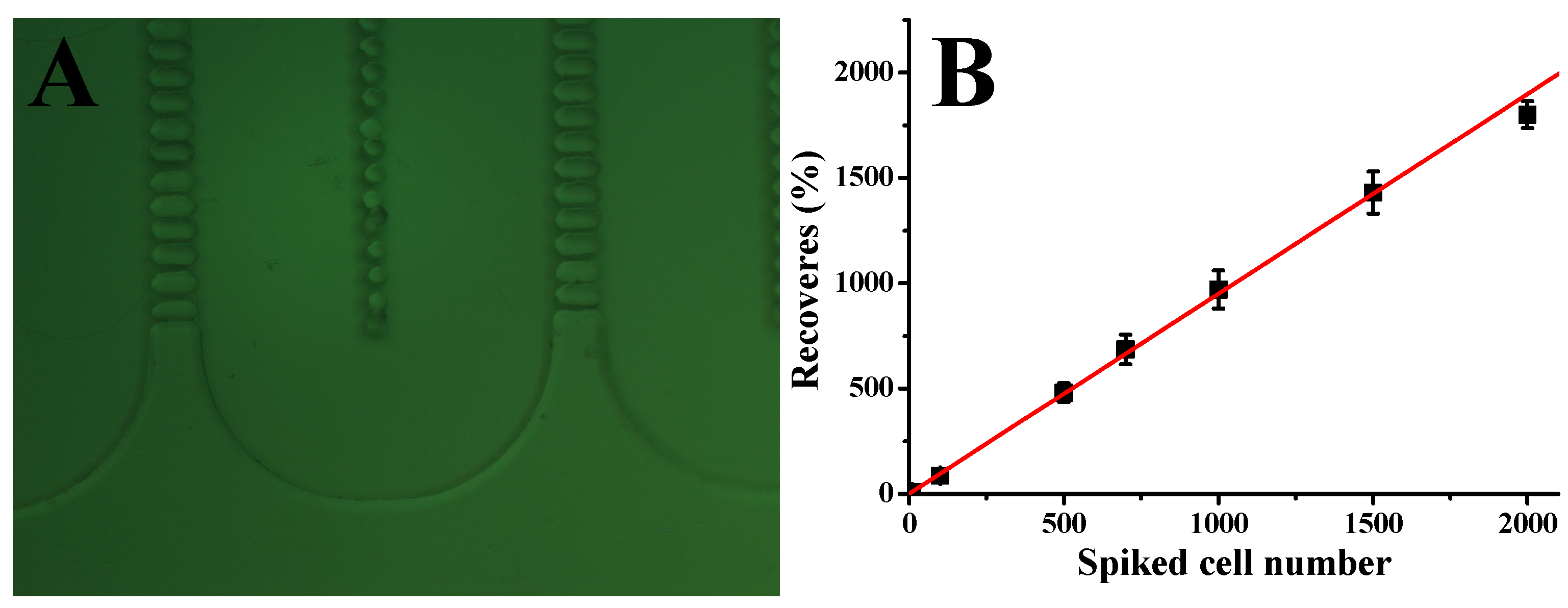
To assess the performance of our CTC capture system in whole human blood, we carried out a series of experiments with blood samples spiked with different concentrations of Hs578t cells under optimized conditions. In particular, 2 mL of whole blood was mixed with Hs578t cells at 10, 50, 100, 500, 1000, 1500, and 2000 cells per mL. The number of cells added to each blood sample was determined using serial dilution, followed by direct cell counting in the spiking volume before the blood was added. These samples were then processed through the microfluidic device at a flow rate of 1.5 µL/s. As shown in
Figure 3B, the system demonstrated a strong linear correlation between the number of spiked cells and the number of cells captured (R
2 = 0.998,
n = 3). The device achieved an average capture efficiency of 90%, indicating consistent performance across a wide range of cell concentrations. These results underscore the reliability and sensitivity of our microfluidic system in isolating CTCs from whole blood, even at low concentrations. The high capture efficiency, combined with the strong linearity of the system, highlights its potential for clinical applications such as early cancer detection, prognosis monitoring, and treatment evaluation. Future studies could further validate the system’s performance in diverse clinical settings and explore its adaptability to other cancer cell types and biomarkers, expanding its applicability in precision medicine.
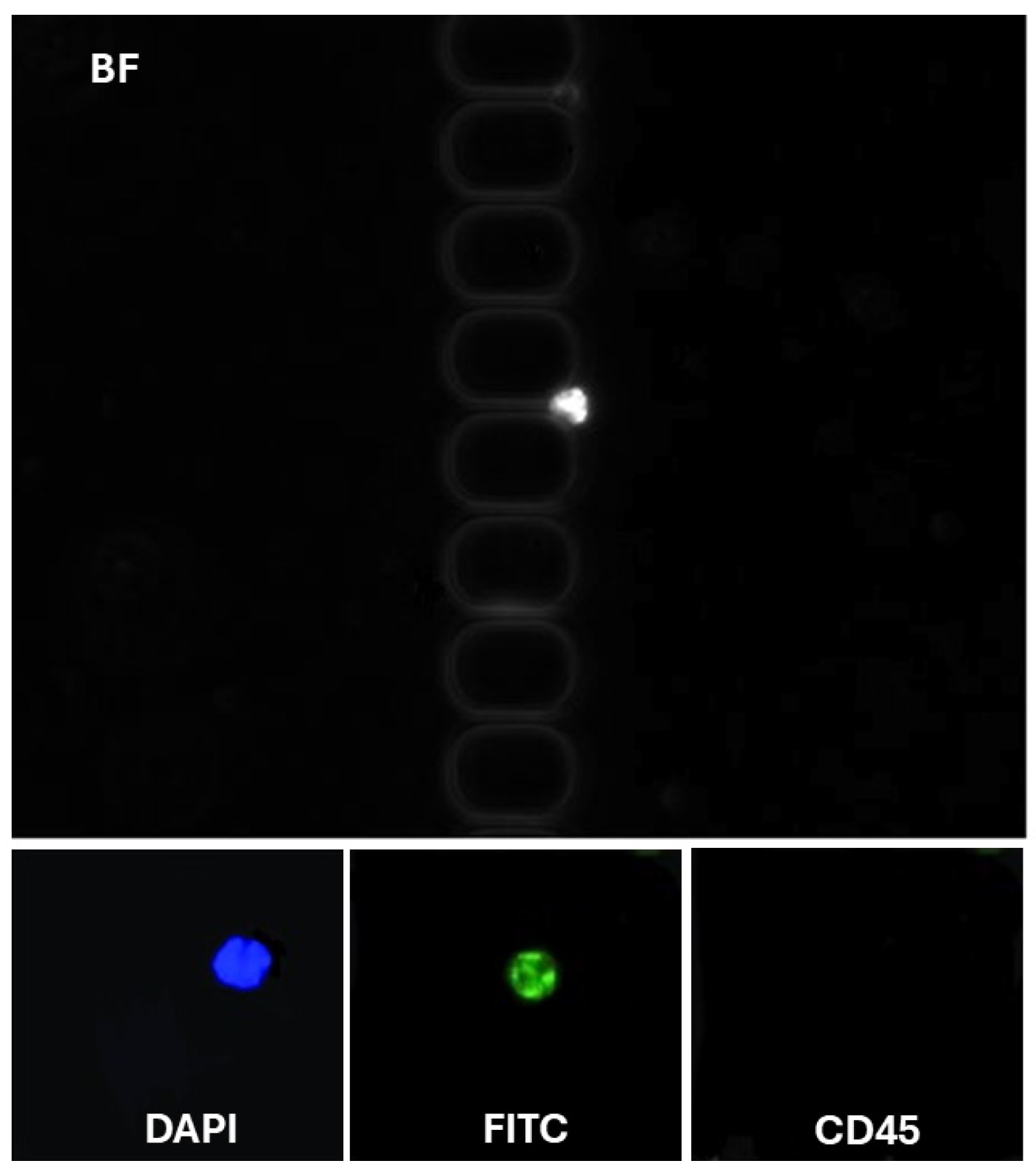
To assess the clinical utility of our methodology, we analyzed blood samples from patients with glioblastoma. To address the challenge of high blood viscosity, 2 mL of blood was diluted with an equal volume of DPBS supplemented with 1% BSA and 1 mM EDTA before being processed using the antibody-functionalized LFA device. The diluted samples were introduced into the device at a controlled flow rate of 1.5 µL/s. Following cell capture, the device underwent a DPBS wash to remove unbound components. Captured cells were fixed with 4% paraformaldehyde (PFA) for 10 min and permeabilized with 0.2% Triton X-100 for another 10 min. Subsequently, the cells were labeled using a mixture of fluorescently tagged antibodies: FITC-conjugated anti-cytokeratin (10 µg/mL), P.E. conjugated anti-CD45 (10 µg/mL), and 500 nM DAPI. This antibody cocktail was introduced into the LFA device and incubated for 30 min, followed by another DPBS wash. CD45 staining was employed to selectively exclude white blood cells from the analysis, as CD45 is a specific marker for leukocytes. Identifying CTCs was based on stringent criteria to ensure specificity and minimize false positives. Cells were classified as CTCs if they exhibited a diameter of at least 9 µm, which aligns with the typically larger size of tumor cells compared to most blood cells [
29]. These cells had to demonstrate positive staining for DAPI
+ and CK-FITC
+, indicative of intact nuclei commonly associated with glioblastoma (CK-FITC
+/DAPI
+/CD45
−) cells classified (
Figure 4). Crucially, these cells were required to lack CD45 expression, confirming their non-leukocyte origin. This multi-marker approach provides a robust framework for distinguishing CTCs from other cellular components in the bloodstream, thereby enhancing the reliability of CTC identification in clinical and research settings. Non-specific signals and debris were excluded from the analysis. On average, 8–12 ± 3 CTCs/mL were detected for each patient (
n = 3). These results validate the enhanced recovery efficiency of our antibody-functionalized LFA device, supported by its ability to isolate EpCAM-positive CTCs with minimal background interference selectively. This approach is promising for advancing liquid biopsy technologies, enabling more precise cancer diagnostics and personalized therapeutic monitoring.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}