
German Porphyria Registry (PoReGer)–Background and Setup

 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Methods
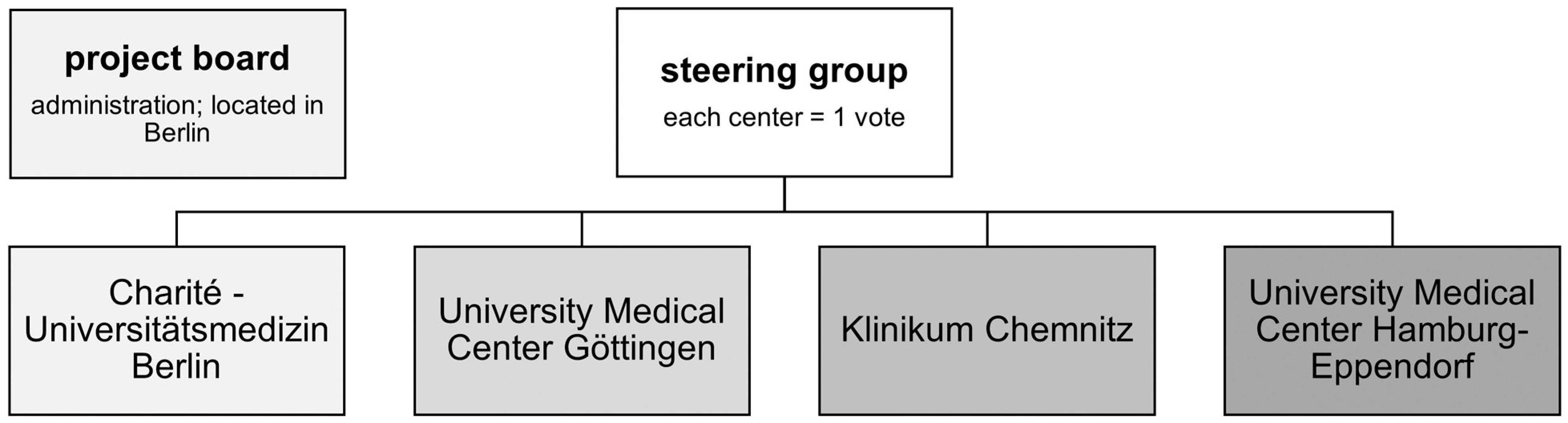
2.1. Founding Centers of the PoReGer-Registry
2.2. Set-Up and Structure of the Registry
2.3. Study Population—Inclusion and Exclusion Criteria
2.4. Ethical Considerations and Data Protection
2.5. Government of PoReGer
3. Discussion
3.1. Founding Centers, Set-Up, Patient Population
3.2. Strengths and Limitations
3.3. Future Plans
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIP | acute intermittent porphyria |
| ALA | aminolevulinic-acid |
| ALADP | 5-aminolevulinic-acid-dehydratase-deficient porphyria |
| AP(s) | acute porphyria(s) |
| CEP | congenital erythropoietic porphyria |
| EPI | European Porphyria Initiative |
| EPP | erythropoietic protoporphyria |
| EPNET | European Porphyria Network |
| EQ-5D-5L | European Quality of Life Questionnaire–5 Dimensions–5 Levels |
| EUCERD | European Union Committee of Experts on Rare Diseases |
| FAS | fatigue assessment scale |
| HCP | hereditary coproporphyria |
| HDP | (erythropoietic) harderoporphyria |
| HEP | hepatoerythropoietic porphyria |
| HIV | humane immunodeficiency virus |
| IPNET | International Porphyria Network |
| NAPOS | Norwegian Porphyria Center |
| PBG | porphobilinogen |
| PCT | porphyria cutanea tarda |
| PHQ-9 | patient health questionnaire–short version with 9 items |
| PoReGer | German Porphyria Registry |
| VP | variegate porphyria |
| XLEPP | X-linked protoporphyria. |
References
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- O’Malley, R.; Rao, G.; Stein, P.; Bandmann, O. Porphyria: Often discussed but too often missed. Pract. Neurol. 2018, 18, 352–358. [Google Scholar] [CrossRef]
- Gouya, L.; Ventura, P.; Balwani, M.; Bissell, D.M.; Rees, D.C.; Stolzel, U.; Phillips, J.D.; Kauppinen, R.; Langendonk, J.G.; Desnick, R.J.; et al. EXPLORE: A Prospective, Multinational, Natural History Study of Patients with Acute Hepatic Porphyria with Recurrent Attacks. Hepatology 2020, 71, 1546–1558. [Google Scholar] [CrossRef]
- Gross, U.; Sassa, S.; Jacob, K.; Deybach, J.C.; Nordmann, Y.; Frank, M.; Doss, M.O. 5-Aminolevulinic acid dehydratase deficiency porphyria: A twenty-year clinical and biochemical follow-up. Clin. Chem. 1998, 44, 1892–1896. [Google Scholar] [CrossRef]
- Frank, J.; Nelson, J.; Wang, X.; Yang, L.; Ahmad, W.; Lam, H.; Jugert, F.K.; Kalka, K.; Poh-Fitzpatrick, M.B.; Goerz, G.; et al. Erythropoietic protoporphyria: Identification of novel mutations in the ferrochelatase gene and comparison of biochemical markers versus molecular analysis as diagnostic strategies. J. Investig. Med. 1999, 47, 278–284. [Google Scholar]
- Kuntz, B.M.; Goerz, G.; Soneborg, H.H.; Lissner, R. HLA-types in porphyria cutanea tarda. Lancet 1981, 1, 155. [Google Scholar] [CrossRef]
- Stein, P.E.; Edel, Y.; Mansour, R.; Mustafa, R.A.; Sandberg, S.; Members of the Acute Porphyria Expert, P. Key terms and definitions in acute porphyrias: Results of an international Delphi consensus led by the European porphyria network. J. Inherit. Metab. Dis. 2023, 46, 662–674. [Google Scholar] [CrossRef]
- Elder, G.; Harper, P.; Badminton, M.; Sandberg, S.; Deybach, J.C. The incidence of inherited porphyrias in Europe. J. Inherit. Metab. Dis. 2013, 36, 849–857. [Google Scholar] [CrossRef]
- Stolzel, U.; Doss, M.O.; Schuppan, D. Clinical Guide and Update on Porphyrias. Gastroenterology 2019, 157, 365–381 e364. [Google Scholar] [CrossRef]
- Singal, A.K. Porphyria cutanea tarda: Recent update. Mol. Genet. Metab. 2019, 128, 271–281. [Google Scholar] [CrossRef]
- Stolzel, U.; Stauch, T.; Kubisch, I. [Porphyria]. Internist 2021, 62, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Dickey, A.K.; Leaf, R.K.; Balwani, M. Update on the Porphyrias. Annu. Rev. Med. 2023, 75. epub ahead of print. [Google Scholar] [CrossRef]
- Lefever, S.; Peersman, N.; Meersseman, W.; Cassiman, D.; Vermeersch, P. Development and validation of diagnostic algorithms for the laboratory diagnosis of porphyrias. J. Inherit. Metab. Dis. 2022, 45, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Gerischer, L.M.; Scheibe, F.; Numann, A.; Kohnlein, M.; Stolzel, U.; Meisel, A. Acute porphyrias—A neurological perspective. Brain Behav. 2021, 11, e2389. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: Pathophysiology, genetics, clinical manifestations, and management. Mol. Genet. Metab. 2019, 128, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Gouya, L.; Malonova, E.; Lamoril, J.; Camadro, J.M.; Flamme, M.; Rose, C.; Lyoumi, S.; Da Silva, V.; Boileau, C.; et al. Mutations in human CPO gene predict clinical expression of either hepatic hereditary coproporphyria or erythropoietic harderoporphyria. Hum. Mol. Genet. 2005, 14, 3089–3098. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Astrin, K.H. Congenital erythropoietic porphyria: Advances in pathogenesis and treatment. Br. J. Haematol. 2002, 117, 779–795. [Google Scholar] [CrossRef]
- Anderson, K.E.; Lobo, R.; Salazar, D.; Schloetter, M.; Spitzer, G.; White, A.L.; Young, R.M.; Bonkovsky, H.L.; Frank, E.L.; Mora, J.; et al. Biochemical Diagnosis of Acute Hepatic Porphyria: Updated Expert Recommendations for Primary Care Physicians. Am. J. Med. Sci. 2021, 362, 113–121. [Google Scholar] [CrossRef]
- Stein, P.; Badminton, M.; Barth, J.; Rees, D.; Stewart, M.F.; British; Irish Porphyria, N. Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann. Clin. Biochem. 2013, 50, 217–223. [Google Scholar] [CrossRef]
- Diehl-Wiesenecker, E.; Blaschke, S.; Wohmann, N.; Kubisch, I.; Stauch, T.; Mainert, M.; Helm, F.; von Wegerer, S.; Pittrow, D.; Frank, J.; et al. Detect Acute Porphyrias in Emergency Departments (DePorED)—A pilot study. Orphanet J. Rare Dis. 2023, 18, 146. [Google Scholar] [CrossRef]
- Ayme, S.; Rodwell, C. The European Union Committee of Experts on Rare Diseases: Three productive years at the service of the rare disease community. Orphanet J. Rare Dis. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Tollanes, M.C.; Aarsand, A.K.; Villanger, J.H.; Stole, E.; Deybach, J.C.; Marsden, J.; To-Figueras, J.; Sandberg, S.; European Porphyria, N. Establishing a network of specialist Porphyria centres—Effects on diagnostic activities and services. Orphanet J. Rare Dis. 2012, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Mykletun, M.; Aarsand, A.K.; Stole, E.; Villanger, J.H.; Tollanes, M.C.; Baravelli, C.; Sandberg, S. Porphyrias in Norway. Tidsskr. Nor. Laegeforen. 2014, 134, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Thomsen, J.; Enes, A.R.; Sandberg, S.; Aarsand, A.K. Health-related quality of life in porphyria cutanea tarda: A cross-sectional registry based study. Health Qual Life Outcomes 2020, 18, 84. [Google Scholar] [CrossRef]
- Thunell, S.; Floderus, Y.; Henrichson, A.; Harper, P. Porphyria in Sweden. Physiol. Res. 2006, 55 (Suppl. 2), S109–S118. [Google Scholar] [CrossRef]
- Linet, M.S.; Gridley, G.; Nyren, O.; Mellemkjaer, L.; Olsen, J.H.; Keehn, S.; Adami, H.O.; Fraumeni, J.F., Jr. Primary liver cancer, other malignancies, and mortality risks following porphyria: A cohort study in Denmark and Sweden. Am. J. Epidemiol. 1999, 149, 1010–1015. [Google Scholar] [CrossRef]
- Kauppinen, R.; Mustajoki, P. Prognosis of acute porphyria: Occurrence of acute attacks, precipitating factors, and associated diseases. Medicine 1992, 71, 1–13. [Google Scholar] [CrossRef]

{kind=link}
| Demographics |
| Age, sex, body mass index |
| social status, education, first digit of postal code, degree of disability |
| Diagnosis |
| Date/year of first manifestation of symptoms and date/year of diagnosis |
| Diagnosis made by which specialty |
| Gene mutation, family history |
| Disease manifestation and clinical course |
| Symptoms at first manifestation and in the further course |
| Number of acute attacks (APs) |
| Chronic symptoms |
| Number and frequency of medical treatment and hospitalizations |
| Trigger factors and measures to prevent them |
| Influence of menstrual cycle/female hormones on symptoms (APs) |
| Investigations |
| Physical examination |
| Laboratory results (general and porphyria specific) |
| Imaging and liver investigations (ultrasound, magnetic resonance imaging (MRI), elastography, liver biopsy) |
| Dermatological examination and skin type (Fitzpatrick-Scale) |
| Neurological examination |
| Results of neurological investigations, if necessary (cranial computed tomography/MRI, nerve conduction studies) |
| Gynecological examination and history |
| Therapies for porphyria |
| Symptomatic and disease modifying treatments/interventions |
| Side effects |
| Medical history |
| comorbidities |
| Medications for comorbidities |
| History of surgical interventions (e.g., appendectomy, gall bladder removal, etc.) |
| History of alcohol consumption, smoking status, illicit drugs |
| Questionnaires |
| Quality of life (EQ-5D-5L) |
| Depression (PHQ-9) |
| Fatigue (FAS) |
| Inclusion Criteria |
|
|
|
| Exclusion Criteria |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerischer, L.; Mainert, M.; Wohmann, N.; Kubisch, I.; Stölzel, U.; Stauch, T.; von Wegerer, S.; Braun, F.; Weiler-Normann, C.; Blaschke, S.; et al. German Porphyria Registry (PoReGer)–Background and Setup. Healthcare 2024, 12, 111. https://doi.org/10.3390/healthcare12010111
Gerischer L, Mainert M, Wohmann N, Kubisch I, Stölzel U, Stauch T, von Wegerer S, Braun F, Weiler-Normann C, Blaschke S, et al. German Porphyria Registry (PoReGer)–Background and Setup. Healthcare. 2024; 12(1):111. https://doi.org/10.3390/healthcare12010111
Chicago/Turabian StyleGerischer, Lea, Mona Mainert, Nils Wohmann, Ilja Kubisch, Ulrich Stölzel, Thomas Stauch, Sabine von Wegerer, Fabian Braun, Christina Weiler-Normann, Sabine Blaschke, and et al. 2024. "German Porphyria Registry (PoReGer)–Background and Setup" Healthcare 12, no. 1: 111. https://doi.org/10.3390/healthcare12010111
APA StyleGerischer, L., Mainert, M., Wohmann, N., Kubisch, I., Stölzel, U., Stauch, T., von Wegerer, S., Braun, F., Weiler-Normann, C., Blaschke, S., Frank, J., Somasundaram, R., & Diehl-Wiesenecker, E. (2024). German Porphyria Registry (PoReGer)–Background and Setup. Healthcare, 12(1), 111. https://doi.org/10.3390/healthcare12010111