
A Proposed Global Medicines Agency (GMA) to Make Biological Drugs Accessible: Starting with the League of Arab States


Abstract
1. Introduction
2. The GMA Charter
- Access and affordability: Individuals and communities face challenges in accessing biotechnology drugs in resource-limited settings. This includes analyzing factors such as cost, availability, distribution, and the role of pharmaceutical companies and government policies;
- Cultural and social dimensions: How biotechnology drugs are perceived, understood, and utilized within specific cultural contexts. This involves exploring local beliefs, practices, and values related to health, illness, and treatment options, as well as the influence of social and cultural factors on the acceptance and use of these drugs;
- Global health disparities: Analyzing the implications of global health disparities in the distribution and availability of biotechnology drugs. They investigate the power dynamics between developed and developing countries, pharmaceutical companies, and regulatory bodies and their impact on access to these medications.
- Ethical considerations: Examining the ethical dimensions of developing, testing, and distributing biotechnology drugs in developing countries. This includes exploring issues of informed consent, clinical trials, and the involvement of vulnerable populations;
- Domestic manufacturing: The cost of establishing the manufacturing of biotechnology drugs keeps developing countries from becoming self-sufficient in their supply. However, this option applies only to those drugs that can be copied in the SRA countries; an example is biosimilars.
- To make these drugs available in developing countries by simplifying the registration process of novel SRA country products;
- To encourage SRA originators and domestic developers by making a large market population with a single registration;
- To ensure data integrity by not requiring all countries to acquire registration dossiers.
- To ensure the safety and efficacy of products manufactured in non-SRA countries by adopting a rational and practical registration process.
2.1. Regulatory Agencies
2.2. Functions of the GMA
- Legally binding registration across member countries, regardless of their geography;
- No dossier sharing with member countries;
- Allows local labeling requirement compliance;
- Does not engage in price negotiations;
- The product must be registered in the country of origin;
- Automatic registration of SRA-approved products if they are also distributed in the country of origin;
- Rapporteur-based evaluation of registration dossiers from non-SRA countries;
- Third-party cGMP audit and testing sample surveillance;
- Centrally operated international scientific, legal, and technical expertise.
3. Finding a Sponsor
3.1. History
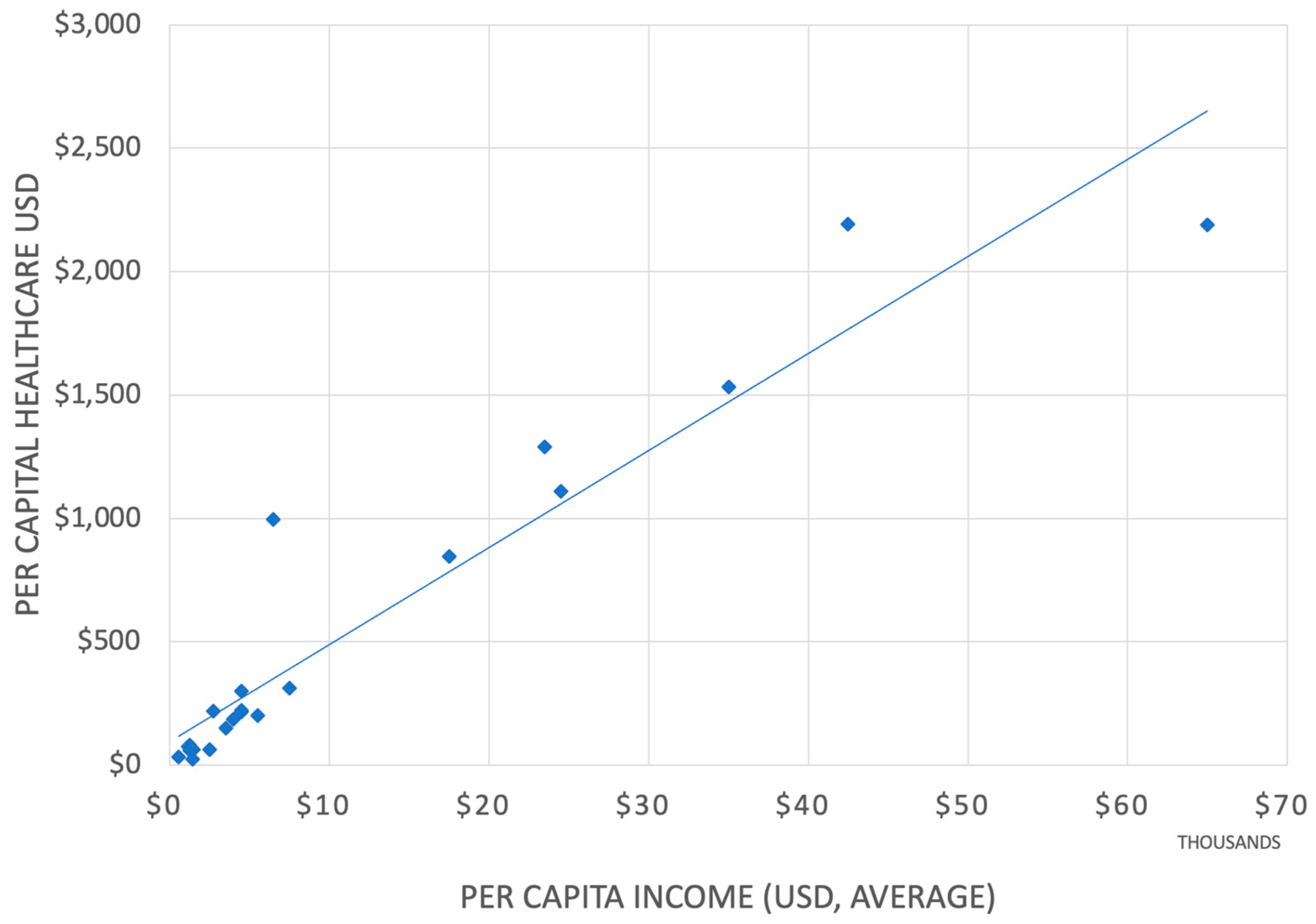
3.2. Affordability
3.3. Markets
4. Regulatory Misconceptions of Arab Countries
- “Regulatory bodies need scientists to evaluate a dossier. Others suggested that the region has plenty of scientists, pharmacists, and academicians to serve this duty”.There is a great misunderstanding among Arab agencies about the qualification of regulatory staff. These include analytical chemists, pharmacokinetics, clinicians, statisticians, quality assurance auditors, lawyers, and others with specific expertise. Unless trained in a particular function, pharmacists, academicians, scientists, and politically appointed heads of the agency should not be part of any dossier evaluation. For this reason, I recommend installing a system of rapporteur evaluation of all biosimilar submissions. In addition, the FDA employs 11,000 full-time scientists [37], so it is not expected of any non-SRI agency to be able to evaluate a regulatory dossier on its own.
- “Discussion between national and international regulatory bodies is needed to ensure biosimilar approval is consistent worldwide”.This is a broader goal that has never been possible, even for generic chemical drugs; there are agencies like the ICH and WHO that provide guidelines that are often adopted, but to expect that regulatory bodies will agree on issues that take their authority away, is not likely to be happy. However, this is what I am suggesting, but with a narrow goal.
- “The limitations faced by recently established regulatory bodies must be recognized and addressed by mature regulatory bodies worldwide”.What is meant by a “mature” authority; an “immature” authority should not operate in the first place. Expecting the FDA or EMA support can only be limited to following their guidelines. Suppose it is meant that the region’s authorities have been operating longer to help the newcomers. In that case, this, too, is misleading, for a more extended operation does not necessarily mean maturity.
- “Countries with greater experience must support countries with less experience of biosimilars”.It is doubtful that any country in the region has the required experience to make them a teacher. Therefore, the right thing to do is to harmonize the registration process, where a single agency approves biosimilars that all member countries will accept.
- “Action should be taken to ensure that all biosimilar products globally are traceable at batch level to ensure adequate pharmacovigilance is upheld. Biosimilar naming will be key to this”.This is not an issue; all products have registration and batch numbers, and the brand naming system is widely accepted to ensure traceability.
- Strong governmental regulators should be in place to ensure drug products can be tracked.Traceability is a fundamental process that applies to all drugs, which is the essential function of any regulatory authority.
- “The long-term effects of switching and multi-switching between biosimilars and/or reference products need to be understood and addressed. This requires a concerted international effort to develop an optimal methodological approach”.This is a misconception; there is no risk in interchangeability that is only an issue in the US, and that, too, is about to be removed. Therefore, there is no need to dwell on this wasteful exercise [38].
- “Biosimilar patient registries could be established and implemented to gather further data on switching”.
- “Electronic healthcare records need to be developed and implemented to facilitate pharmacovigilance and gather further data on switching”.Member countries can’t have electronic health records; pharmacovigilance is a common practice for all drugs. So, there is no need to extend this to switching.
- “Encourage meeting with clinicians to explain what biosimilars are and what they are not, to enable them how to decide whether to prescribe biosimilars”.Clinicians are least qualified to understand the nuances of the regulatory process; they must trust agencies’ decisions. No need to waste time teaching clinicians.
- “Physicians, pharmacists, regulators, patients, and all stakeholders must communicate and share their experiences—challenges, and successes—with biosimilars”.This wasteful exercise is more of a slogan; there is no need to teach or promote biosimilars. All must accept an approved biosimilar; promoting its safety and efficacy may even cause doubt about its safety and efficacy.
- Interestingly, except for Iran, which is not part of the Arab countries, all other countries in the Arab countries require clinical efficacy testing [32]. In addition, all Arab World regulatory authorities follow the FDA or EMA, and Egypt also includes the WHO [41].The WHO is not a regulatory agency; it is an Agency whose decisions are widely criticized since it operates mostly on common consensus. The FDA and EMA guidelines depend on many legislative issues and legal exposure and are slow to change the guidelines as new science teaches otherwise.
- A lack of agreement exists among the Arab countries regarding regulatory approval issues, particularly regarding interchangeability and switching. In Saudi Arabia, biosimilars are not automatically interchangeable. For example, ten biosimilars have been approved, of which only two are regarded as interchangeable. It is evident that in Saudi Arabia, biosimilarity alone is not sufficient for substitution or switching. However, biosimilars approved by the EMA are considered interchangeable. Additionally, a clinical trial that involves switching must be run to approve switching, which must happen before the biosimilar is approved.None of these considerations are needed; as allowed in the EU, all biosimilars are interchangeable, even one biosimilar with another. The issue of interchangeability is indigenous to the US and is up for removal.
- The Egyptian Drug Authority (EDA) “Guideline for registration of Biosimilar products in Egypt” is in place as of March 2020. The applicant must exhibit and compare the biosimilarity of their product to the innovator/reference product by completing and comparing pre-clinical and clinical studies and quality exercises. The EDA adopts the EMA guidelines and refers to the U.S. FDA’s safety and quality considerations, the WHO guidelines for evaluating similar biotherapeutic products, and relevant ICH (The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use; https://ich.org/ (accessed on 14 July 2023) guidelines. In Egypt, however, the Ministry of Health will make interchangeability decisions, where the patient will not be given a choice.Misconceptions regarding blanket following lead to archaic animal toxicology testing and other quality assessments that may not be necessary, as described below. An agency should create its guideline, albeit borrowing from any additional guideline, instead of listing another as the marker. Comments for interchangeability apply as stated above.
- The Jordanian FDA’s guidelines are based on the EMA, where the EMA model has been implemented for quality assessment and comparability. It also authorizes the approval of manufacturing sites as a prerequisite to product approval and filing. Currently, six products have been approved according to Jordan’s biosimilar guidelines. Jordan’s approach to biosimilar regulation can be considered vigilant and strict. Nonetheless, biosimilars manufactured and marketed in reference countries, including but not limited to the UK, USA, Germany, France, the Netherlands, Sweden, Australia, Austria, and Japan, are usually given more privileges.The same comments as offered for the Egyptian guideline apply here. In addition, references to biosimilars from some SRA countries should be expanded and automatically allowed registration without reviewing the dossier.
- Medicines in Tunisia are obtained by centralized pharmacy purchase (PCP). The Biosimilar Specialized Committee makes decisions on a case-by-case basis regarding interchangeability. The committee comprises representatives of pharmaceutical inspection, a national control laboratory, regulatory authorities, various clinicians, and experts who utilize biosimilars.The interchangeability issue is redundant; the approval committee need not include anyone who is not qualified to judge the compliance of a dossier. Moreover, approval should not be based on consensus, a significant weakness in almost all Arab state agencies, as elaborated above.
5. Biosimilars
6. Proposed Guideline for Biosimilars
6.1. SRA Sourcing
6.2. Non-SRA Country Biosimilars
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- What Are “Biologics” Questions and Answers. Available online: https://www.fda.gov/about-fda/center-biologics-evaluation-and-research-cber/what-are-biologics-questions-and-answers (accessed on 14 July 2023).
- Von Schwerin, A.; Stoff, H.; Wahrig, B. (Eds.) Biologics: An introduction. In Biologics, a History of Agents Made from Living Organisms in the Twentieth Century; Pickering & Chatto: London, UK, 2013; pp. 1–33. [Google Scholar]
- Gross, C.P.; Sepkowitz, K.A. The myth of the medical breakthrough: Smallpox, vaccination, and Jenner reconsidered. Int. J. Infect. Dis. 1998, 3, 54–60. [Google Scholar] [CrossRef] [PubMed]
- CooperDavid, N.; ReichardtJürgen, K.V.; AliBassam, R.; PatrinosGeorge, P. Which Strategies Should Developing Countries Employ to Invest in Precision Medicine? A New “Fast-Second Winner” Strategy. OMICS J. Integr. Biol. 2017, 21, 647–657. [Google Scholar] [CrossRef]
- Spök, A.; Twyman, R.M.; Fischer, R.; Ma, J.K.; Sparrow, P.A. Evolution of a regulatory framework for pharmaceuticals derived from genetically modified plants. Trends Biotechnol. 2008, 26, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, A.K.; Palaz, F.; Sofija, S.; Elmousa, N.; Ledezma, Y.; Cachat, E.; Rios-Solis, L. Improving recombinant protein production in CHO cells using the CRISPR-Cas system. Biotechnol. Adv. 2023, 64, 108115. [Google Scholar] [CrossRef]
- Global Spending on Medicines in 2010, 2022, and a Forecast for 2027. Available online: https://www.statista.com/statistics/280572/medicine-spending-worldwide/ (accessed on 14 July 2023).
- Andrews, L.; Ralston, S.; Blomme, E.; Barnhart, K. A snapshot of biologic drug development: Challenges and opportunities. Hum. Exp. Toxicol. 2015, 34, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, N.; Flossmann, O.; Hunt, D. Evolution of biological agents: How established drugs can become less safe. BMJ 2017, 357, j1707. [Google Scholar] [CrossRef]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009–2018. JAMA 2020, 323, 844–853, Erratum in: JAMA 2022, 328, 1110. Erratum in: JAMA 2022, 328, 1111. [Google Scholar] [CrossRef]
- Schlander, M.; Hernandez-Villafuerte, K.; Cheng, C.-Y.; Mestre-Ferrandiz, J.; Baumann, M. How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. Pharmacoeconomics 2021, 39, 1243–1269. [Google Scholar] [CrossRef]
- Priciest Drugs in 2023. Available online: https://www.fiercepharma.com/special-reports/priciest-drugs-2023 (accessed on 14 July 2023).
- Kronfol, N.M. Historical development of health systems in the Arab countries: A review. East. Mediterr. Health J. 2012, 18, 1151–1156. Available online: https://apps.who.int/iris/bitstream/handle/10665/118493/EMHJ_2012_18_11_1151_1156.pdf;jsessionid=A6C226BFF2849E550596A7F50848641E?sequence=1 (accessed on 14 July 2023). [CrossRef]
- McCall, S.J.; Semaan, A.; Altijani, N.; Opondo, C.; Abdel-Fattah, M.; Kabakian-Khasholian, T. Trends, wealth inequalities and the role of the private sector in caesarean section in the Middle East and North Africa: A repeat cross-sectional analysis of population-based surveys. PLoS ONE 2021, 16, e0259791. [Google Scholar] [CrossRef]
- Goffman, L.F. Medicine and Health in the Modern Middle East and North Africa. 2019. Available online: https://www.jadaliyya.com/Details/40332 (accessed on 14 July 2023).
- Mate, K.; Bryan, C.; Deen, N.; McCall, J. Review of Health Systems of the Middle East and North Africa Region. In International Encyclopedia of Public Health; Elsevier: Singapore, 2017; pp. 347–356. [Google Scholar] [CrossRef]
- The Middle East: A Pharma Market in the Making. Available online: https://www.reutersevents.com/pharma/commercial/middle-east-pharma-market-making#:~:text=The%20total%20regional%20market%20is,imported%20generics%20and%20branded%20drugs (accessed on 14 July 2023).
- Advancement and Inequity in the Arab World’s Medical and Pharmaceutical Sectors. Available online: https://arabcenterdc.org/resource/advancement-and-inequity-in-the-arab-worlds-medical-and-pharmaceutical-sectors/ (accessed on 14 July 2023).
- Arab Center. Healthcare Resources. Available online: https://arabcenterdc.org/resource/health-care-in-the-arab-world-outcomes-of-a-broken-social-contract/ (accessed on 14 July 2023).
- Arab Language International Council. Available online: https://alarabiah.org/ (accessed on 14 July 2023).
- World Bank. Current Healthcare Expenditure. Available online: https://data.worldbank.org/indicator/sh.xpd.chex.gd.zs (accessed on 14 July 2023).
- Pharma Solutions. Growth of Pharmaceutical Distributors in the Middle East. Available online: https://www.pharmasolutions-int.com/rise-growth-of-pharma-distributors-in-the-middle-east/ (accessed on 14 July 2023).
- Balkhi, B.; Alshayban, D.; Alotaibi, N.M. Impact of Healthcare Expenditures on Healthcare Outcomes in the Middle East and North Africa (MENA) Region: A Cross-Country Comparison, 1995–2015. Front. Public Health 2021, 8, 624962. [Google Scholar] [CrossRef]
- WHO Call for Consultant on Monoclonal Antibodies for Infectious Diseases. Available online: https://www.who.int/news-room/articles-detail/call-for-consultant-on-monoclonal-antibodies-for-infectious-diseases#:~:text=The%20cost%20of%20goods%20to,US%2095%2D200%20per%20gram (accessed on 14 July 2023).
- FDA Continuous Manufacturing. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q13-continuous-manufacturing-drug-substances-and-drug-products (accessed on 14 July 2023).
- Coheren Market Insight of Biological Markets. Available online: https://www.coherentmarketinsights.com/market-insight/mena-biologics-and-biosimilars-market-4393#:~:text=The%20ArabWorld%20biologics%20%26%20biosimilars%20market,period%20(2020%2D2027) (accessed on 14 July 2023).
- Coheren. Market Insight Saudia Arabia Pharmaceuticals. Available online: https://www.coherentmarketinsights.com/market-insight/saudi-arabia-pharmaceuticals-market-3557 (accessed on 14 July 2023).
- Vision 2030 Saudi Arabia. Available online: https://www.vision2030.gov.sa/ (accessed on 14 July 2023).
- Tunisian Investment. Pharmaceutical Sector. Available online: https://tia.gov.tn/storage/app/media/argumentaires/tia_tunisia_pharma/ag%20pharma%20ang.pdf (accessed on 14 July 2023).
- Jordan Times. Jordan Pharmaceutical Exports. Available online: https://www.jordantimes.com/news/local/jordans-pharmaceutical-exports-reach-jd1b-%E2%80%94-japm (accessed on 14 July 2023).
- Market Data Forecast. Middle East Biosimilars Market. Available online: https://www.marketdataforecast.com/market-reports/mea-biosimilars-market (accessed on 14 July 2023).
- Bassil, N.; Sasmaz, S.; Sayah, M.E.; Akalankam, A.; Realizing Biosimilar Potential in the Middle East & Africa. The Middle East and Africa Perspective White Paper [Internet]. IQVIA; 2020 [cited August 24, 2022]: 28. Available online: https://www.iqvia.com/-/media/iqvia/pdfs/mea/white-paper/biosimilar_iqvia-whitepaper_final.pdf (accessed on 20 September 2022).
- RefWorld. Charter of Arab League. Available online: https://www.refworld.org/docid/3ae6b3ab18.html (accessed on 14 July 2023).
- SFDA. Saudi Arabia Hosts 1st Meeting of Arab Authorities Controlling Medicines. Available online: https://www.sfda.gov.sa/en/news/88233 (accessed on 14 July 2023).
- GABI Journal. Stakeholders Meeting. Available online: http://gabi-journal.net/2nd-mena-stakeholder-meeting-on-biosimilars-2018-report.html (accessed on 14 July 2023).
- FDA. FDA Science Forum. Available online: https://www.fda.gov/science-research/about-science-research-fda/fda-science-forum (accessed on 14 July 2023).
- Press Release. Lee Seeks Increated Competition in Biological Drugs. Available online: https://www.lee.senate.gov/2023/7/lee-seeks-increased-competition-in-biological-drug-market (accessed on 14 July 2023).
- EMA. Interchangeable Biosimilar Medicines. Available online: https://www.ema.europa.eu/en/news/biosimilar-medicines-can-be-interchanged (accessed on 14 July 2023).
- Niazi, S.K. Biosimilars: Harmonizing the Approval Guidelines. Biologics 2022, 2, 171–195. [Google Scholar] [CrossRef]
- De Mora, F. Biosimilars: A Value Proposition. Biodrugs 2019, 33, 353–356. [Google Scholar] [CrossRef]
- Niazi, S.K. Molecular Biosimilarity—An AI-Driven Paradigm Shift. Int. J. Mol. Sci. 2022, 23, 10690. [Google Scholar] [CrossRef]
- Beygmoradi, A.; Homaei, A.; Hemmati, R.; Fernandes, P. Recombinant protein expression: Challenges in production and folding related matters. Int. J. Biol. Macromol. 2023, 233, 123407. [Google Scholar] [CrossRef] [PubMed]
- EMA Centrally Approved Biosimilars. Available online: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine/field_ema_med_status/authorised-36/ema_medicine_types/field_ema_med_biosimilar/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar (accessed on 8 June 2022).
- Inxight Drug Database. Available online: https://drugs.ncats.io/substances?facet=Development%20Status%2FUS%20Approved%20Rx&facet=Substance%20Class%2Fprotein&facet=Substance%20Form%2FPrincipal%20Form&page=1 (accessed on 25 April 2023).
- WHO Guidelines on Evaluation of Biosimilars. Replacement of Annex 2 of WHO Technical Report Series, No. 977. Available online: https://www.who.int/publications/m/item/guidelines-on-evaluation-of-biosimilars (accessed on 8 June 2022).
- Niazi, S. The WHO Biosimilar Guidance Is Based on Weak Science. Available online: https://www.centerforbiosimilars.com/view/who-biosimilar-guidance-is-based-on-weak-science (accessed on 14 July 2023).
- Niazi, S. Opinion: One Step Forward, Half Step Back. Available online: https://www.centerforbiosimilars.com/view/opiniononestepforwardhalfastepbackforwhobiosimilarguidance (accessed on 8 June 2022).
- CDSCO, India. Available online: https://cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadAlertsFiles/BiosimilarGuideline2016.pdf (accessed on 8 June 2022).
- FDA Biosimilar Labeling. Available online: https://www.fda.gov/files/drugs/published/Labeling-for-Biosimilar-Products-Guidance-for-Industry.pdf (accessed on 14 July 2023).
- Forbes Magazine One Man’s Mission to Fix the FDA’s Biosimilars Problem. Available online: https://www.forbes.com/sites/nicolefisher/2018/07/25/one-mans-mission-to-fix-the-fdas-biosimilar-problem/?sh=2386ed9d2380 (accessed on 14 July 2023).
- Niazi, S.K. End animal testing for biosimilar approval. Science 2022, 377, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Florian, J.; Campbell, E.; Schrieber, S.J.; Bai, J.P.; Weaver, J.L.; Hyland, P.L.; Thway, T.M.; Matta, M.K.; Lankapalli, R.H.; et al. Advancing Biosimilar Development Using Pharmacodynamic Biomarkers in Clinical Pharmacology Studies. Clin. Pharmacol. Ther. 2019, 107, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S. Scientific Rationale for Waiving Clinical Efficacy Testing of Biosimilars. Drug Des. Dev. Ther. 2022, 16, 2803–2815. [Google Scholar] [CrossRef]
- MHRA. Biosimilar Guidance. Available online: https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products (accessed on 8 June 2022).
- Global Biosimilar Guidelines. Available online: https://www.gabionline.net/reports/Guidelines-for-biosimilars-around-the-world (accessed on 14 July 2023).
- García-Arieta, A.; Simon, C.; Tam, A.; Santos, G.M.L.; Fernandes, E.A.F.; Martínez, Z.R.; Rodrigues, C.; Park, S.A.; Kim, J.Y.; Kim, K.; et al. A Survey of the Regulatory Requirements for the Waiver of In Vivo Bioequivalence Studies of Generic Products in Certain Dosage Forms by Participating Regulators and Organisations of the International Pharmaceutical Regulators Programme. J. Pharm. Pharm. Sci. 2021, 24, 113–126. [Google Scholar] [CrossRef]
- Niazi, S.K. The Coming of Age of Biosimilars: A Personal Perspective. Biologics 2022, 2, 107–127. [Google Scholar] [CrossRef]
- Niazi, S.K. Biosimilars: A futuristic fast-to-market advice to developers. Expert Opin. Biol. Ther. 2021, 22, 149–155. [Google Scholar] [CrossRef] [PubMed]
- FDA. Real World Evidence Program. Available online: https://www.fda.gov/drugs/development-resources/advancing-real-world-evidence-program (accessed on 14 July 2023).
- Chen, Y.; Monnard, A.; Jorge Santos Da, S. An Inflection Point for Biosimilars, McKinsey & Co. [cited 2021 Oct 12]. Available online: https://www.mckinsey.com/industries/life-sciences/our-insights/an-inflection-point-for-biosimilars (accessed on 8 June 2022).
- US Senate. The FDA Modernization Act 2.0. Available online: https://www.congress.gov/bill/117th-congress/senate-bill/5002 (accessed on 14 July 2023).
- FDA. Applied Regulatory Science. Available online: https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/division-applied-regulatory-science (accessed on 14 July 2023).
- Chiu, K.; Racz, R.; Burkhart, K.; Florian, J.; Ford, K.; Garcia, M.I.; Geiger, R.M.; Howard, K.E.; Hyland, P.L.; Ismaiel, O.A.; et al. New science, drug regulation, and emergent public health issues: The work of FDA’s division of applied regulatory science. Front. Med. 2023, 9, 1109541. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. FDA Guidance: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product. 2016. Available online: https://www.fda.gov/media/88622/download (accessed on 14 July 2023).
- Li, L.; Ma, L.; Schrieber, S.J.; Rahman, N.A.; Deisseroth, A.; Farrell, A.T.; Wang, Y.; Sinha, V.; Marathe, A. Quantitative Relationship between AUEC of Absolute Neutrophil Count and Duration of Severe Neutropenia for G-CSF in Breast Cancer Patients. Clin. Pharmacol. Ther. 2018, 104, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Sheikhy, M.; Schrieber, S.J.; Sun, Q.; Gershuny, V.; Matta, M.K.; Bai, J.P.; Du, X.; Vegesna, G.; Shah, A.; Prentice, K.; et al. Considerations for Use of Pharmacodynamic Biomarkers to Support Biosimilar Development—(I) A Randomized Trial with PCSK9 Inhibitors. Clin. Pharmacol. Ther. 2022, 113, 71–79. [Google Scholar] [CrossRef]
- Gershuny, V.; Sun, Q.; Schrieber, S.J.; Matta, M.K.; Weaver, J.L.; Ji, P.; Sheikhy, M.; Hsiao, C.; Vegesna, G.; Shah, A.; et al. Considerations for Use of Pharmacodynamic Biomarkers to Support Biosimilar Development—(II) A Randomized Trial with IL-5 Antagonists. Clin. Pharmacol. Ther. 2022, 113, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Florian, J.; Gershuny, V.; Sun, Q.; Schrieber, S.J.; Matta, M.K.; Hazel, A.; Sheikhy, M.; Weaver, J.L.; Hyland, P.L.; Hsiao, C.; et al. Considerations for Use of Pharmacodynamic Biomarkers to Support Biosimilar Development—(III) A Randomized Trial with Interferon Beta-1a Products. Clin. Pharmacol. Ther. 2022, 113, 339–348. [Google Scholar] [CrossRef]
- Hyland, P.L.; Chekka, L.M.S.; Samarth, D.P.; Rosenzweig, B.A.; Decker, E.; Mohamed, E.G.; Guo, Y.; Matta, M.K.; Sun, Q.; Wheeler, W.; et al. Evaluating the Utility of Proteomics for the Identification of Circulating Pharmacodynamic Biomarkers of IFNβ-1a Biologics. Clin. Pharmacol. Ther. 2022, 113, 98–107. [Google Scholar] [CrossRef]
- Niazi, S.K. Functional Biosimilarity to Replace Clinical Efficacy Testing of mAb Biosimilars: Advancing the FDA Perspective. Preprints.org 2023, 2023041141. [Google Scholar] [CrossRef]
- Woodcock. Available online: https://www.biopharmadive.com/news/fdas-woodcock-the-clinical-trial-system-is-broken/542698/ (accessed on 14 July 2023).
- US Congress. Available online: https://www.govinfo.gov/content/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdf (accessed on 14 July 2023).
- FDA. Biosimilars Science and Research. Available online: https://www.fda.gov/drugs/biosimilars/biosimilars-science-and-research (accessed on 14 July 2023).
- EMA Biosimilar EPAR Program. Available online: https://www.ema.europa.eu/en/search/search/field_ema_web_categories%253Aname_field/Human/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar?search_api_views_fulltext=epar%20biosimilar (accessed on 14 July 2023).
- FDA. Biosimilars Approval Documents. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 14 July 2023).
- Clinical Trial Database. Available online: https://clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=With&Search=Apply (accessed on 14 July 2023).
- Biosimilar Clinical Trials @PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/?term=biosimilar+clinical+trial (accessed on 14 July 2023).
- US Food and Drug Administration. FDA Draft Guidance: Biomarker Qualification: Evidentiary Framework. 2018. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM628118.pdf (accessed on 25 April 2023).
- Florian, J.; Sun, Q.; Schrieber, S.J.; White, R.; Shubow, S.; Johnson-Williams, B.E.; Sheikhy, M.; Harrison, N.R.; Parker, V.J.; Wang, Y.; et al. Pharmacodynamic Biomarkers for Biosimilar Development and Approval: A Workshop Summary. Clin. Pharmacol. Ther. 2022, 113, 1030–1035. [Google Scholar] [CrossRef]
- Sharma, V.K.; Patapoff, T.W.; Kabakoff, B.; Pai, S.; Hilario, E.; Zhang, B.; LI, C.; Borisov, O.; Kelley, R.F.; Chorny, I.; et al. In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. USA 2014, 111, 18601–18606. [Google Scholar] [CrossRef]
- Cymera, F.; Becka, H.; Rohde, A.; Reusch, D. Therapeutic monoclonal antibody N-glycosylation—Structure, function and therapeutic potential. Biologicals 2018, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.; Hufton, S.E.; Fox, B.; Dougall, T.; Rigsby, P.; Bristow, A.; Participants of the Study. International standards for monoclonal antibodies to support pre- and post-marketing product consistency: Evaluation of a candidate international standard for the bioactivities of rituximab. mAbs 2017, 10, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Ryding, J.; Stahl, M.; Ullmann, M. Demonstrating biosimilar and originator antidrug antibody binding comparability in antidrug antibody assays: A practical approach. Bioanalysis 2017, 9, 1395–1406. [Google Scholar] [CrossRef]
- Wang, X.; An, Z.; Luo, W.; Xia, N.; Zhao, Q. Molecular and functional analysis of monoclonal antibodies in support of biologics development. Protein Cell 2017, 9, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Generally Accepted Scientific Knowledge in Applications for Drug and Biological Products: Nonclinical Information. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/generally-accepted-scientific-knowledge-applications-drug-and-biological-products-nonclinical (accessed on 14 July 2023).
- Wang, Y.; Strauss, D.G. Advancing Innovations in Biosimilars. Clin. Pharmacol. Ther. 2022, 113, 11–15. [Google Scholar] [CrossRef]
- Strauss, D.G.; Wang, Y.; Florian, J.; Zineh, I. Pharmacodynamic Biomarkers Evidentiary Considerations for Biosimilar Development and Approval. Clin. Pharmacol. Ther. 2022, 113, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hötzel, I.; Theil, F.P.; Bernstein, L.J.; Prabhu, S.; Deng, R.; Quintana, L.; Lutman, J.; Sibia, R.; Chan, P.; Buymbaca, D.; et al. A strategy for risk mitigation of antibodies with fast clearance. In MAbs; Taylor & Francis: Abingdon, UK, 2012; Volume 4, pp. 753–760. [Google Scholar]
- FDA. Generally Accepted Scientific Knowledge in Applications for Drug and Biological Products: Nonclinical Information Guidance for Industry. Available online: https://www.fda.gov/media/168408/download (accessed on 14 July 2023).
- US Senate. Biological Price Competition and Innovation Act. Available online: https://www.fda.gov/media/78946/download (accessed on 14 July 2023).
- Code of Federal Register Title 21. Bioavailability and Bioequivalence. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=320.25 (accessed on 14 July 2023).
- World Health Organization. WHO/List of Stringent Regulatory Authorities (SRAs); World Health Organization: Geneva, Switzerland, 2022; Available online: https://www.who.int/initiatives/who-listed-authority-reg-authorities/SRAs (accessed on 14 July 2023).
- World Health Organization; WHO Expert Committee on Specifications for Pharmaceutical Preparations. Fifty-Second Report of the WHO Expert Committee on Specifications for Pharmaceutical Preparations; World Health Organization: Geneva, Switzerland, 2018; pp. 355–356. ISBN 978-92-4-121019-5. OCLC 1039407367. [Google Scholar]
- Outlook India. Biocon Bribery Case. Available online: https://www.outlookindia.com/business/biocon-biologics-biocon-biologics-bribery-cbi-arrests-joint-drug-controller-for-alleged-bribery-to-favour-biocon-biologics-medicine-news-203753 (accessed on 14 July 2023).
- Financier World. Corruption in the Pharmaceutical Sector. Available online: https://www.financierworldwide.com/bribery-and-corruption-in-the-pharmaceutical-sector#.Y1AWiOzMKlM (accessed on 14 July 2023).
- FDA. Data integrity and Compliance with cGMP. Available online: https://www.fda.gov/files/drugs/published/Data-Integrity-and-Compliance-With-Current-Good-Manufacturing-Practice-Guidance-for-Industry.pdf (accessed on 14 July 2023).
- Barbier, L.; Mbuaki, A.; Simoens, S.; Declerck, P.; Vulto, A.G.; Huys, I. Regulatory Information and Guidance on Biosimilars and Their Use Across Europe: A Call for Strengthened One Voice Messaging. Front. Med. 2022, 9, 820755. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. No two classes of biosimilars: Urgent advice to the US Congress and the FDA. J. Clin. Pharm. Ther. 2022, 47, 1352–1361. [Google Scholar] [CrossRef]
- African Vaccine Manufacturing Initiative. Establishing Manufacturing Capabilities for Human Vaccines. Available online: https://www.avmi-africa.org/projects/current-projects/#1513773628836-0f0598a8-ec17 (accessed on 14 July 2023).


{kind=link}
| Rank | Drug | Manufacturers | Indication | Cost |
|---|---|---|---|---|
| 1 | Hemgenix | CSL Behring, uniQure | Hemophilia B | $3.5 million/dose |
| 2 | Skysona | Bluebird bio | Cerebral adrenoleukodystrophy | $3 million/dose |
| 3 | Zynteglo | Bluebird bio | Transfusion-dependent thalassemia | $2.8 million/dose |
| 4 | Zolgensma | Novartis | Spinal muscular atrophy | $2.25 million/dose |
| 5 | Myalept | Chiesi Farmaceutici | Leptin deficiency | $1.26 million/year |
| 6 | Zokinvy | Eiger BioPharmaceuticals | Hutchinson-Gilford progeria syndrome and processing-deficient progeroid laminopathies | $1.07 million/year |
| 7 | Danyelza | Y-mAbs Therapeutics | Relapsed or refractory high-risk neuroblastoma | $1.01 million/year |
| 8 | Kimmtrak | Immunocore | Uveal melanoma | $0.97 million/year |
| 9 | Luxturna | Spark Therapeutics | Biallelic RPE65-mediated inherited retinal disease | $0.85 million/treatment |
| 10 | Folotyn | Acrotech Biopharma | Relapsed or refractory peripheral T-cell lymphoma | $0.84 million/year |
| Drug Name | 2022 Sales, USD Billion |
|---|---|
| Actemra/RoActemra (tocilizumab) | USD 2.58 |
| Darzalex (daratumumab) | USD 7.98 |
| Dupixent (dupilumab) | USD 17.42 |
| Enbrel (etanercept) | USD 4.12 |
| Eylea (aflibercept) | USD 12.72 |
| Hemlibra (emicizumab) | USD 3.65 |
| Humira (adalimumab) | USD 21.24 |
| Imfinzi (durvalumab) | USD 2.78 |
| Lantus (insulin glargine) | USD 2.38 |
| Ocrevus (ocrelizumab) | USD 5.76 |
| Opdivo (nivolumab) | USD 8.25 |
| Perjeta (pertuzumab) | USD 3.90 |
| Prolia (denosumab) | USD 3.63 |
| Remicade (infliximab) | USD 2.34 |
| Skyrizi (risankizumab) | USD 5.17 |
| Stelara (ustekinumab) | USD 9.72 |
| Taltz (ixekizumab) | USD 2.48 |
| Tecentriq (atezolizumab) | USD 3.55 |
| Tremfya (guselkumab) | USD 2.67 |
| Trulicity (dulaglutide) | USD 7.44 |
| Drug | Patent Expiry |
|---|---|
| Interferon beta-1b | 2004 |
| Parathyroid hormone | 2004 |
| Interferon alfa-2b | 2004 |
| Chorionic gonadotropin | 2007 |
| Interferon alfa-n3 | 2011 |
| Etanercept | 2012 |
| Menotropins | 2015 |
| Urofollitropin | 2015 |
| Peginterferon alfa-2b | 2015 |
| Interferon beta-1a | 2020 |
| Insulin regular | 2025 |
| Insulin lispro | 2014 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K. A Proposed Global Medicines Agency (GMA) to Make Biological Drugs Accessible: Starting with the League of Arab States. Healthcare 2023, 11, 2075. https://doi.org/10.3390/healthcare11142075
Niazi SK. A Proposed Global Medicines Agency (GMA) to Make Biological Drugs Accessible: Starting with the League of Arab States. Healthcare. 2023; 11(14):2075. https://doi.org/10.3390/healthcare11142075
Chicago/Turabian StyleNiazi, Sarfaraz K. 2023. "A Proposed Global Medicines Agency (GMA) to Make Biological Drugs Accessible: Starting with the League of Arab States" Healthcare 11, no. 14: 2075. https://doi.org/10.3390/healthcare11142075
APA StyleNiazi, S. K. (2023). A Proposed Global Medicines Agency (GMA) to Make Biological Drugs Accessible: Starting with the League of Arab States. Healthcare, 11(14), 2075. https://doi.org/10.3390/healthcare11142075