
Alzheimer Identification through DNA Methylation and Artificial Intelligence Techniques


Abstract
:1. Introduction
1.1. Forecasting and Classification Models
1.2. CpG DNA Methylation
1.3. Paper Structure
2. Literature Review
3. Materials and Methods
3.1. Initial Filtering
- estimate a linear regression with Y as the dependent variable. Save the p-value for each .
- Filter off the with (p-value) .with .
3.2. Main Algorithm
- Create a vector grid (D) with the each component representing the dimension (group of ) includes in the simulation. Two grids are included, a fine grid with relative small differences in the values of the elements (representing the dimensions that the researcher considers more likely) and a broad grid with large differences in values.The values inside the above grids represent the selected. As an example, represents . and are the constant step increases in the fine and broad grids, respectively. For instance, and are the second and third elements in the fine grid. The actual elements related to this second and third values depend on the actual value of . If then the second and third elements related to and , respectively, while if , then they relate to and , respectively. Where , each of these values, i.e., is the number of chosen. is a constant that specifies (together with ) the total size of the fine grid, while is the analogous term for the broad grid. For simplicity purposes the case of a fine grid, starting a , followed by a broad grid has been shown but this is not a required constraint. The intent is giving discretion to the researcher to apply the fine grid to the area that is considered more important. This is an attempt to bring the expertise of the researcher into the algorithm. In Equation (12) it can be seen the combination of these two grids (D).
- Create a mapping between each , where each is a vector, and 10 decile regions. The group of with the highest of the p-value are included in the first decile and assigned a probability of . The group of with the second highest of the p-value are included in the second decile and assigned a probability of . This process is repeated for all deciles creating a mapping.where B is a vector of probabilities. In this way, the with the largest p-values are more likely to be included.
- For each generate , i=1,...,m, a random number with . If then is not included in the preliminary group of . Otherwise it is included. In this way a filtering is carried out.
- Randomly elements of are chosen.
- Estimate the Hit Ratio (HR)where TE is the total number of classification estimations and CE is the number of correct classification estimates.
- Repeat steps (3) to (6) k times for each . In this way there is a mapping:
- 7.
- Define new search interval between the two highest success rates:Iteration 1 (Iter = 1) ends, identifying interval:
- 8.
- Divide the interval identified in the previous step into steps.where and
- 9.
- Create a new mapping estimating the new hit rates (following the same approach as in previous steps)
- 10.
- Repeat times until the maximum number of iterations () is reached.or until the desire hit rate () is reachedor until no further HR improvement is achieved. Select .
3.3. Data
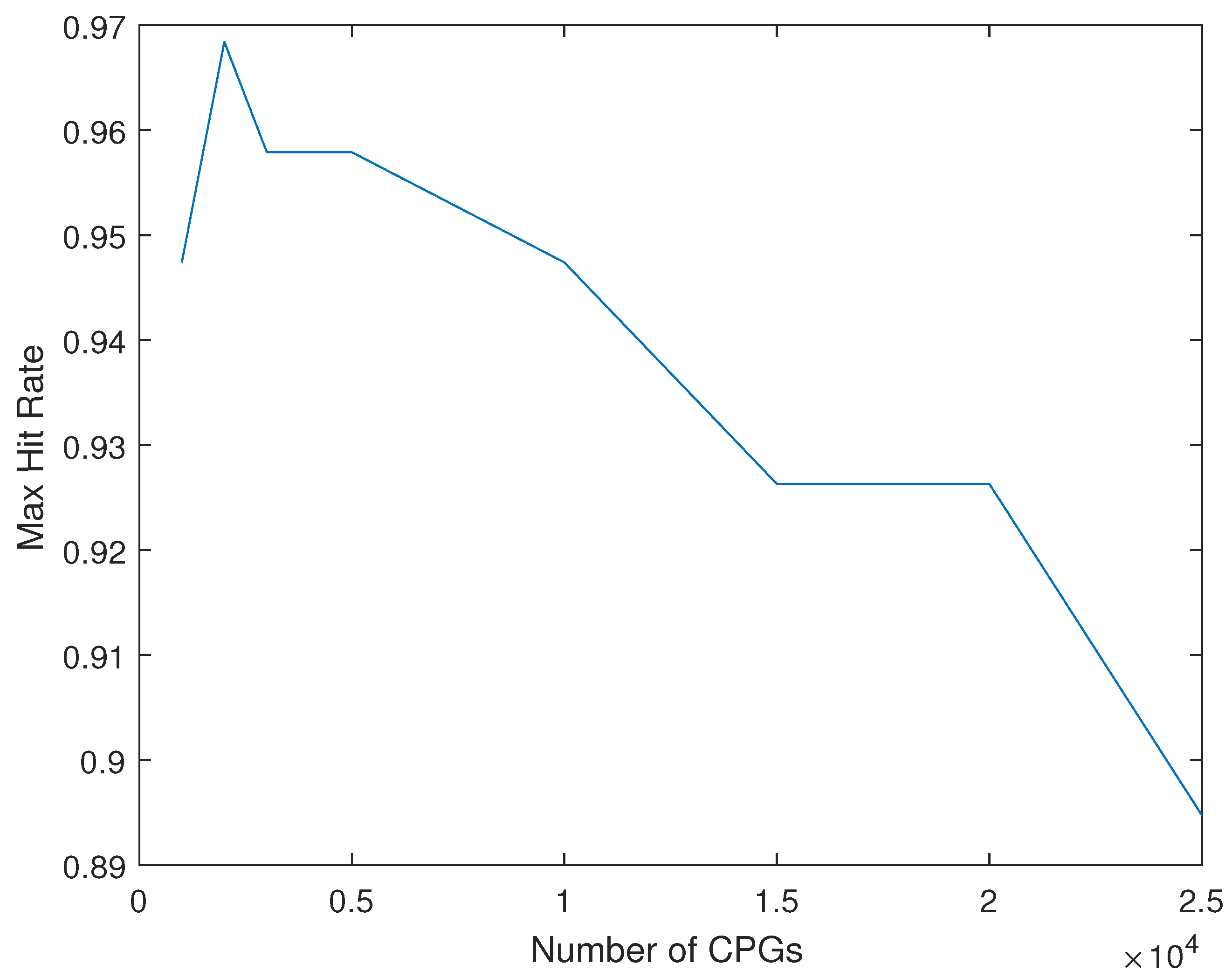
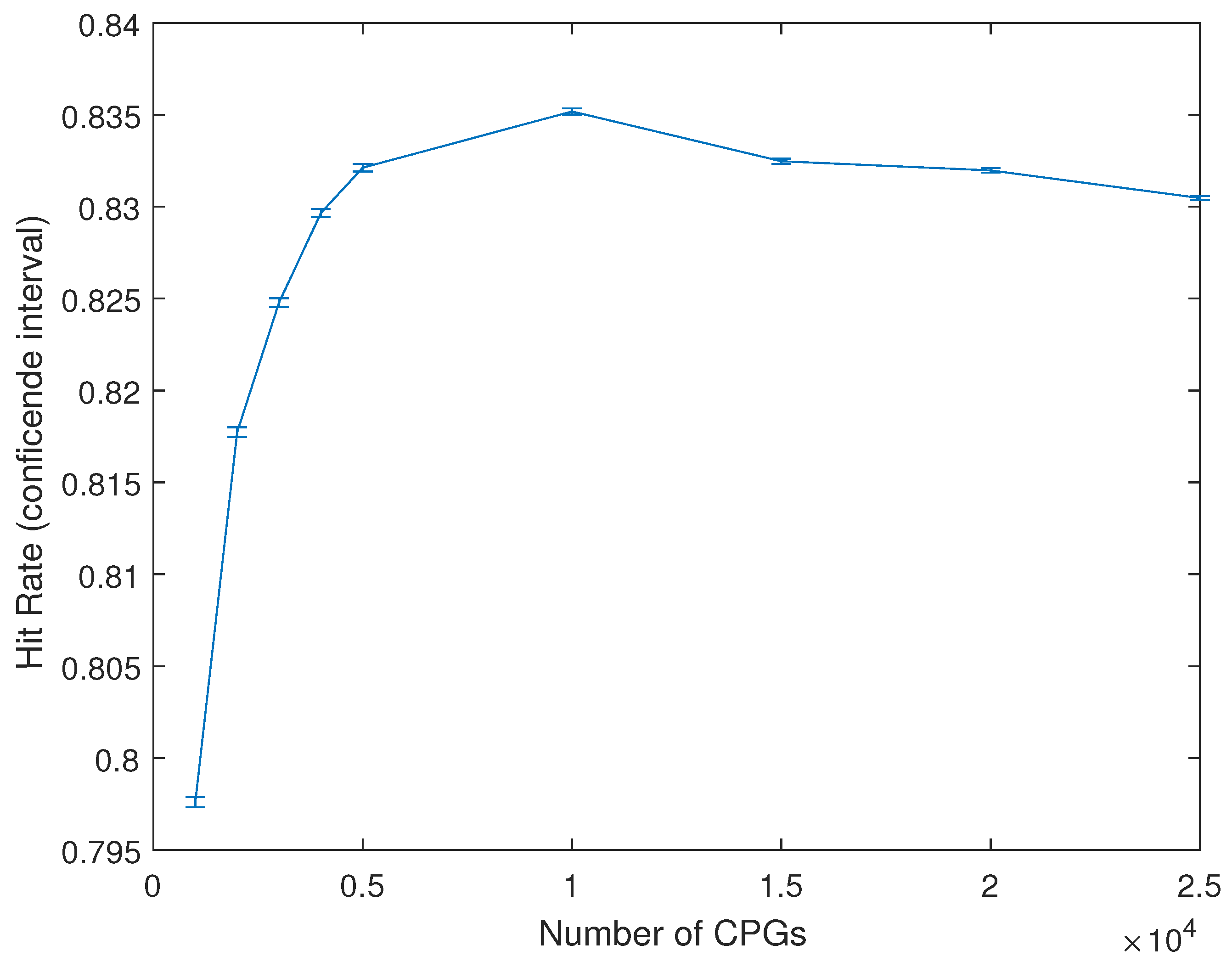
4. Results
4.1. Single Data Set
4.2. Multiple Data Sets
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Olivari, B.S.; Baumgart, M.; Taylor, C.A.; McGuire, L.C. Population measures of subjective cognitive decline: A means of advancing public health policy to address cognitive health. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12142. [Google Scholar]
- Donohue, M.C.; Sperling, R.A.; Salmon, D.P.; Rentz, D.M.; Raman, R.; Thomas, R.G.; Weiner, M.; Aisen, P.S. The preclinical Alzheimer cognitive composite: Measuring amyloid-related decline. JAMA Neurol. 2014, 71, 961–970. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.G.; Kopelman, M.D. The memory deficits in Alzheimer-type dementia: A review. Q. J. Exp. Psychol. 1986, 38, 575–602. [Google Scholar] [CrossRef]
- Greene, J.D.; Hodges, J.R.; Baddeley, A.D. Autobiographical memory and executive function in early dementia of Alzheimer type. Neuropsychologia 1995, 33, 1647–1670. [Google Scholar] [CrossRef]
- Sahakian, B.J.; Morris, R.G.; Evenden, J.L.; Heald, A.; Levy, R.; Philpot, M.; Robbins, T.W. A comparative study of visuospatial memory and learning in Alzheimer-type dementia and Parkinson’s disease. Brain 1988, 111, 695–718. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- Hsieh, C.L. Dependence of transcriptional repression on CpG methylation density. Mol. Cell. Biol. 1994, 14, 5487–5494. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.N.; Krawczak, M. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum. Genet. 1989, 83, 181–188. [Google Scholar] [CrossRef]
- Vertino, P.M.; Yen, R.; Gao, J.; Baylin, S.B. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol. Cell. Biol. 1996, 16, 4555–4565. [Google Scholar] [CrossRef] [Green Version]
- Gudjonsson, J.E.; Krueger, G. A role for epigenetics in psoriasis: Methylated cytosine–guanine sites differentiate lesional from nonlesional skin and from normal skin. J. Investig. Dermatol. 2012, 132, 506–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornélie, S.; Wiel, E.; Lund, N.; Lebuffe, G.; Vendeville, C.; Riveau, G.; Vallet, B.; Ban, E. Cytosine-phosphate-guanine (CpG) motifs are sensitizing agents for lipopolysaccharide in toxic shock model. Intensive Care Med. 2002, 28, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Wong, C.C.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef] [Green Version]
- Marabita, F.; Almgren, M.; Lindholm, M.E.; Ruhrmann, S.; Fagerström-Billai, F.; Jagodic, M.; Sundberg, C.J.; Ekström, T.J.; Teschendorff, A.E.; Tegnér, J.; et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics 2013, 8, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Kuan, P.F.; Wang, S.; Zhou, X.; Chu, H. A statistical framework for Illumina DNA methylation arrays. Bioinformatics 2010, 26, 2849–2855. [Google Scholar] [CrossRef]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-mediated epigenetic reprograming enhances anti-leukemia efficacy of CD123-targeted chimeric antigen receptor T-cells. Front. Immunol. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Rhee, I.; Jair, K.W.; Yen, R.W.C.; Lengauer, C.; Herman, J.G.; Kinzler, K.W.; Vogelstein, B.; Baylin, S.B.; Schuebel, K.E. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000, 404, 1003–1007. [Google Scholar] [CrossRef]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.K.; Hsu, H.S.; Chang, J.W.; Chen, C.Y.; Chen, J.T.; Wang, Y.C. Alteration of DNA methyltransferases contributes to 5 CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007, 55, 205–213. [Google Scholar] [CrossRef]
- Haupt, S.E.; Cowie, J.; Linden, S.; McCandless, T.; Kosovic, B.; Alessandrini, S. Machine learning for applied weather prediction. In Proceedings of the 2018 IEEE 14th International Conference on e-Science (e-Science), Amsterdam, The Netherlands, 29 October–1 November 2018; pp. 276–277. [Google Scholar]
- Stefanovič, P.; Štrimaitis, R.; Kurasova, O. Prediction of flight time deviation for lithuanian airports using supervised machine learning model. Comput. Intell. Neurosci. 2020. [Google Scholar] [CrossRef]
- Rafiee, P.; Mirjalily, G. Distributed Network Coding-Aware Routing Protocol Incorporating Fuzzy-Logic-Based Forwarders in Wireless Ad hoc Networks. J. Netw. Syst. Manag. 2020, 28, 1279–1315. [Google Scholar] [CrossRef]
- Roshani, M.; Phan, G.; Roshani, G.H.; Hanus, R.; Nazemi, B.; Corniani, E.; Nazemi, E. Combination of X-ray tube and GMDH neural network as a nondestructive and potential technique for measuring characteristics of gas-oil–water three phase flows. Measurement 2021, 168, 108427. [Google Scholar] [CrossRef]
- Pourbemany, J.; Essa, A.; Zhu, Y. Real Time Video based Heart and Respiration Rate Monitoring. arXiv 2021, arXiv:2106.02669. [Google Scholar]
- Alfonso, G.; Carnerero, A.D.; Ramirez, D.R.; Alamo, T. Stock forecasting using local data. IEEE Access 2020, 9, 9334–9344. [Google Scholar] [CrossRef]
- Joachims, T. SVM-Light: Support Vector Machine, version 6.02; University of Dortmund: Dortmund, Germany, 1999. [Google Scholar]
- Meyer, D.; Leisch, F.; Hornik, K. The support vector machine under test. Neurocomputing 2003, 55, 169–186. [Google Scholar] [CrossRef]
- Wang, L. Support Vector Machines: Theory and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2005; Volume 177. [Google Scholar]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Sung, E. A study of AdaBoost with SVM based weak learners. In Proceedings of the 2005 IEEE International Joint Conference on Neural Networks, Montreal, QC, Canada, 31 July–4 August 2005; Volume 1, pp. 196–201. [Google Scholar]
- Magnin, B.; Mesrob, L.; Kinkingnéhun, S.; Pélégrini-Issac, M.; Colliot, O.; Sarazin, M.; Dubois, B.; Lehéricy, S.; Benali, H. Support vector machine-based classification of Alzheimer’s disease from whole-brain anatomical MRI. Neuroradiology 2009, 51, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lu, S.; Dong, Z.; Yang, J.; Yang, M.; Zhang, Y. Dual-tree complex wavelet transform and twin support vector machine for pathological brain detection. Appl. Sci. 2016, 6, 169. [Google Scholar] [CrossRef] [Green Version]
- Fetahu, I.S.; Ma, D.; Rabidou, K.; Argueta, C.; Smith, M.; Liu, H.; Wu, F.; Shi, Y.G. Epigenetic signatures of methylated DNA cytosine in Alzheimer’s disease. Sci. Adv. 2019, 5, eaaw2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tost, J. DNA methylation: An introduction to the biology and the disease-associated changes of a promising biomarker. Mol. Biotechnol. 2010, 44, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.A.; Wang, Z.; Wu, X.; Kernstine, K.H.; Riggs, A.D.; Pfeifer, G.P. DNA methylation biomarkers for lung cancer. Tumor Biol. 2012, 33, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Vidaki, A.; Ballard, D.; Aliferi, A.; Miller, T.H.; Barron, L.P.; Court, D.S. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci. Int. Genet. 2017, 28, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossi, E.; Stoccoro, A.; Tannorella, P.; Migliore, L.; Coppedè, F. Artificial neural networks link one-carbon metabolism to gene-promoter methylation in Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 53, 1517–1522. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Ha, J.; Park, S. Prediction of Alzheimer’s disease based on deep neural network by integrating gene expression and DNA methylation dataset. Expert Syst. Appl. 2020, 140, 112873. [Google Scholar] [CrossRef]
- Bhasin, M.; Reinherz, E.L.; Reche, P.A. Recognition and classification of histones using support vector machine. J. Comput. Biol. 2006, 13, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Liu, H.; Zheng, Y.; He, Y.; Lu, D.; Lyu, C. A reliable method for colorectal cancer prediction based on feature selection and support vector machine. Med. Biol. Eng. Comput. 2019, 57, 901–912. [Google Scholar] [CrossRef]
- Lalkhen, A.G.; McCluskey, A. Clinical tests: Sensitivity and specificity. Contin. Educ. Anaesth. Crit. Care Pain 2008, 8, 221–223. [Google Scholar] [CrossRef] [Green Version]
- Tanzi, R.E. FDA Approval of Aduhelm Paves a New Path for Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 2714–2715. [Google Scholar] [CrossRef] [PubMed]
- Karlawish, J.; Grill, J.D. The approval of Aduhelm risks eroding public trust in Alzheimer research and the FDA. Nat. Rev. Neurol. 2021, 17, 523–524. [Google Scholar] [CrossRef]
- Ayton, S. Brain volume loss due to donanemab. Eur. J. Neurol. 2021, 28, e67–e68. [Google Scholar] [CrossRef]
- Vellas, B.J. The Geriatrician, the Primary Care Physician, Aducanumap and the FDA Decision: From Frustration to New Hope. J. Nutr. Health Aging 2021, 25, 821–823. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| GEO Code | Cases | Tissue | Illness |
|---|---|---|---|
| GSE66351 | 190 | Glian and neuron | AD and control |
| GSE80970 | 286 | Pre-frontal cortex and gyrus | AD and control |
| Number | CpG (Indetifier) | Methylation Level |
|---|---|---|
| 1 | cg13869341 | 0.89345 |
| 2 | cg14008030 | 0.71088 |
| … | … | … |
| 481,868 | cg05999368 | 0.51372 |
| AD/Control | 0 |
| Number | CpG (Indetifier) | Methylation Level |
|---|---|---|
| 2 | cg14008030 | 0.71088 |
| 481,868 | cg05999368 | 0.51372 |
| AD/Control | 0 |
| Patient 1 | Patient 2 | Patient 3 |
|---|---|---|
| 0.71088 | 0.63174 | 0.72582 |
| 0.51372 | 0.62145 | 0.43212 |
| 0 | 1 | 0 |
| Patient 1 | Patient 2 | Patient 3 |
|---|---|---|
| … | … | … |
| Controls | HR (Linear) | HR (Gaussian) | HR (Polynomial) | CpGs |
|---|---|---|---|---|
| None | 0.8211 | 0.7921 | 0.8167 | All |
| Age | 0.8947 | 0.8142 | 0.8391 | All |
| Gender | 0.8211 | 0.7921 | 0.8167 | All |
| Cell type | 0.8211 | 0.7921 | 0.8167 | All |
| Brain Region | 0.8211 | 0.7921 | 0.8167 | All |
| Controls | Hit Rate | CpGs |
|---|---|---|
| None | 0.7263 | 41,784 |
| Age | 0.8424 | 41,784 |
| Gender | 0.7263 | 41,784 |
| Cell type | 0.7263 | 41,784 |
| Brain Region | 0.7263 | 41,784 |
| Controls | Hit Rate | CpGs |
|---|---|---|
| GSE66351 | 0.8710 | 4300 |
| GSE80970 | 0.9517 | 4300 |
| All | 0.9202 | 4300 |
| Ratio | All | GSE66351 | GSE80970 |
|---|---|---|---|
| Sensitivity | 0.9007 | 0.8333 | 0.9506 |
| Specificity | 0.9485 | 0.9394 | 0.9531 |
| PPV | 0.9621 | 0.9615 | 0.9625 |
| NPV | 0.8679 | 0.7561 | 0.9385 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfonso Perez, G.; Caballero Villarraso, J. Alzheimer Identification through DNA Methylation and Artificial Intelligence Techniques. Mathematics 2021, 9, 2482. https://doi.org/10.3390/math9192482
Alfonso Perez G, Caballero Villarraso J. Alzheimer Identification through DNA Methylation and Artificial Intelligence Techniques. Mathematics. 2021; 9(19):2482. https://doi.org/10.3390/math9192482
Chicago/Turabian StyleAlfonso Perez, Gerardo, and Javier Caballero Villarraso. 2021. "Alzheimer Identification through DNA Methylation and Artificial Intelligence Techniques" Mathematics 9, no. 19: 2482. https://doi.org/10.3390/math9192482
APA StyleAlfonso Perez, G., & Caballero Villarraso, J. (2021). Alzheimer Identification through DNA Methylation and Artificial Intelligence Techniques. Mathematics, 9(19), 2482. https://doi.org/10.3390/math9192482