
Applied Proteomics in ‘One Health’



 and
and 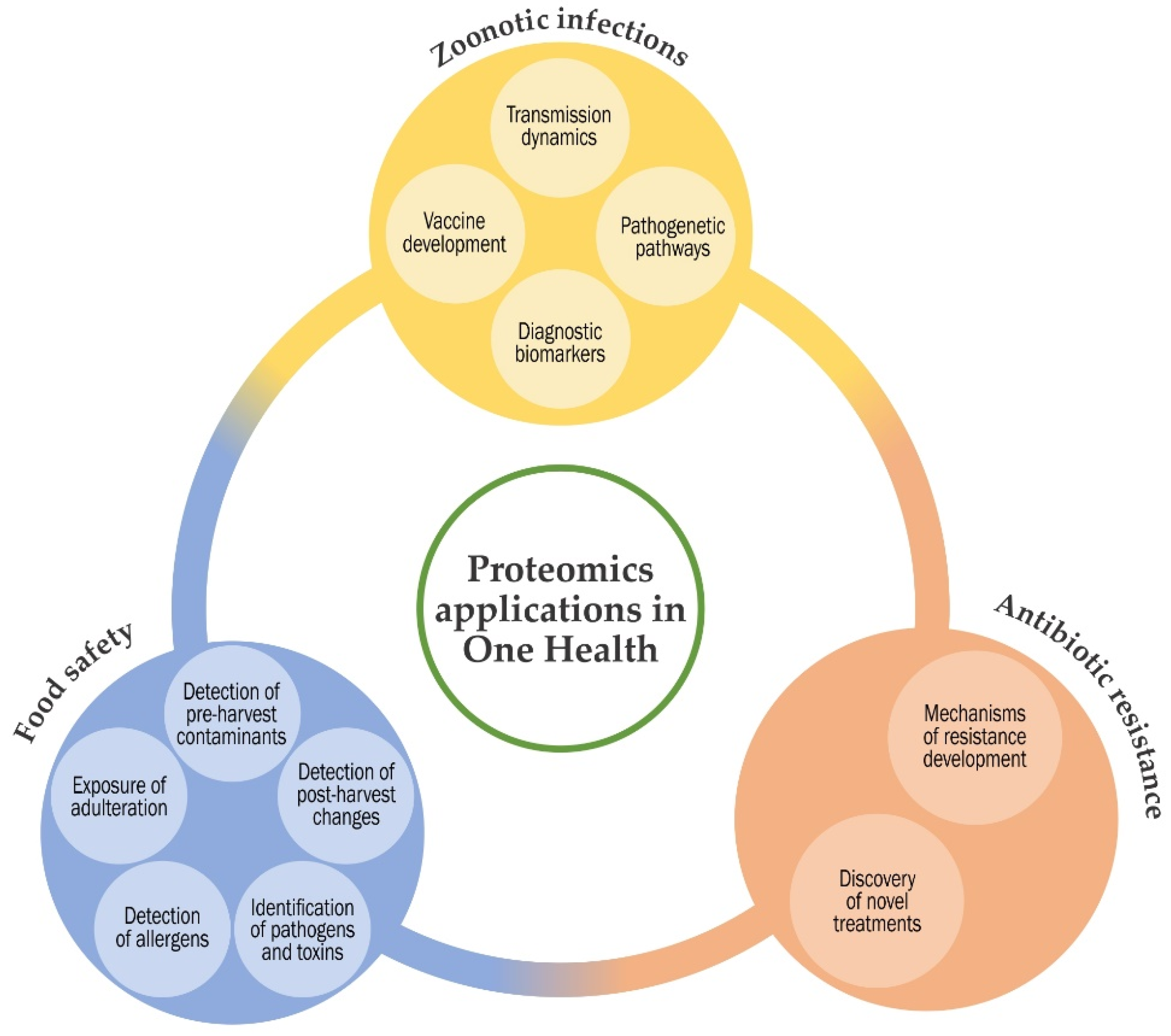{kind=link}
Abstract
1. Introduction
2. Proteomics Applications in Zoonotic Infections
2.1. Prion Zoonotic Diseases
2.2. Viral Zoonotic Infections
2.2.1. Rabies
2.2.2. Coronavirus Infections
2.2.3. West Nile Fever
2.2.4. Influenza Virus Infections
2.3. Bacterial Zoonotic Infections
2.3.1. Mycobacterium avium subsp. Paratuberculosis Infection
2.3.2. Mycobacterium Bovis Infection
2.3.3. Listeria monocytogenes Infections
2.3.4. Bartonella henselae Infections
2.3.5. Brucella spp. Infections
2.3.6. Burkholderia mallei Infections
2.3.7. Campylobacter spp. Infections
2.3.8. Coxiella burnetii Infections
2.3.9. Francicella tularensis Infections
2.3.10. Salmonella spp. Infections
2.3.11. Borrelia spp. Infections
2.3.12. Leptospira spp. Infections
2.3.13. Other Infections
2.4. Protozoan Zoonotic Infections
2.4.1. Cryptosporidium parvum Infections
2.4.2. Toxoplasma gondii Infections
2.4.3. Giardia duodenalis Infections
2.4.4. Leishmania Infections
2.5. Metazoan Zoonotic Infections
2.5.1. Ancylostoma caninum Infections
2.5.2. Angiostrongylus cantonensis Infections
2.5.3. Trichinella spiralis spp. Infections
2.5.4. Echinococcus spp. Infections
3. Proteomics Applications in Antibiotic Resistance
4. Proteomics Applications in Food Safety
4.1. Detection of Pre-Harvest Contaminants and Post-Harvest Changes
4.2. Study of Product Traits
4.3. Identification of Pathogens and Toxins
4.4. Detection of Allergens
4.5. Exposure of Adulteration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Organisation for Animal Health 2021. Available online: https://www.oie.int (accessed on 1 April 2021).
- Marco-Ramell, A.; de Almeida, A.M.; Cristobal, S.; Rodrigues, P.; Roncada, P.; Bassols, A. Proteomics and the search for welfare and stress biomarkers in animal production in the one-health context. Mol. Biosyst. 2016, 12, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Pasquali, C.; Appel, R.D.; Ou, K.; Golaz, O.; Sanchez, J.C.; Yan, J.X.; Gooley, A.A.; Hughes, G.; Humphery-Smith, I.; et al. From proteins to proteomes: Large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology 1996, 14, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Görg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 2000, 21, 1037–1053. [Google Scholar] [CrossRef]
- Görg, A.; Drews, O.; Lück, C.; Weiland, F.; Weiss, W. 2-DE with IPGs. Electrophoresis 2009, 30 (Suppl. 1), S122–S132. [Google Scholar] [CrossRef]
- Abdallah, C.; Dumas-Gaudot, E.; Renaut, J.; Sergeant, K. Gel-based and gel-free quantitative proteomics approaches at a glance. Int. J. Plant Genom. 2012, 2012, 494572. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Lelong, C.; Rabilloud, T. What room for two-dimensional gel-based proteomics in a shotgun proteomics world? Proteomes 2020, 8, 17. [Google Scholar] [CrossRef]
- Millioni, R.; Franchinb, C.; Tessaria, P.; Polati, R.; Cecconid, D.; Arrigoni, G. Pros and cons of peptide isolectric focusing in shotgun proteomics. J. Chromatogr. A 2013, 1293, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fornelli, L.; Toby, T.K.; Schachner, L.F.; Doubleday, P.F.; Srzentić, K.; DeHart, C.J.; Kelleher, N.L. Top-down proteomics: Where we are, where we are going? J. Proteom. 2018, 175, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Palagi, P.M.; Hernandez, P.; Walther, D.; Appel, R.D. Proteome informatics I: Bioinformatics tools for processing experimental data. Proteomics 2006, 6, 5435–5444. [Google Scholar] [CrossRef] [PubMed]
- Anagnostopoulos, A.K.; Stravopodis, D.J.; Tsangaris, G.T. Yield of 6000 proteins by 1D nLC-MS/MS without pre-fractionation. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1047, 92–96. [Google Scholar] [CrossRef]
- Katsafadou, A.I.; Fthenakis, G.C.; Anagnostopoulos, A.K.; Barbagianni, M.S.; Spanos, S.A.; Mavrogianni, V.S.; Billinis, C.; Tsangaris, G.T. Proteomic analysis of blood and milk of ewes with Mannheimia haemolytica mastitis. In Proceedings of the 13th Greek Veterinary Congress, Athens, Greece, 8–10 May 2015; p. 37. [Google Scholar]
- Katsafadou, A.I. Proteomic Study of Ovine Mastitis associated with Mannheimia haemolytica. Ph.D. Thesis, University of Thessaly, Karditsa, Greece, 2017. [Google Scholar]
- Centers for Disease Controls and Prevention. Prion Diseases. Available online: https://www.cdc.gov/prions/index.html (accessed on 31 May 2021).
- Bauerfeind, R.; von Graevenitz, A.; Kimmig, P.; Schiefer, H.G.; Schwarz, T.; Slenczka, W.; Zahner, H. Zoonoses, Infectious Diseases Transmissible from Animals to Humans, 4th ed.; ASM Press: Washington, DC, USA, 2016. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Stahl, N.; Baldwin, M.A.; Teplow, D.B.; Hood, L.; Gibson, B.W.; Burlingame, A.L.; Prusiner, S.B. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 1993, 32, 1991–2002. [Google Scholar] [CrossRef]
- Turk, E.; Teplow, D.B.; Hood, L.E.; Prusiner, S.B. Purification and properties of the cellular and scrapie hamster prion proteins. Eur. J. Biochem. 1988, 176, 21–30. [Google Scholar] [CrossRef]
- Graham, J.F.; Kurian, D.; Agarwal, S.; Toovey, L.; Hunt, L.; Kirby, L.; Pinheiro, T.J.; Banner, S.J.; Gill, A.C. Na+/K+-ATPase is present in scrapie-associated fibrils, modulates PrP misfolding in vitro and links PrP function and dysfunction. PLoS ONE 2011, 6, e26813. [Google Scholar] [CrossRef]
- Batxelli-Molina, I.; Salvetat, N.; Andréoletti, O.; Guerrier, L.; Vicat, G.; Molina, F.; Mourton-Gilles, C. Ovine serum biomarkers of early and late phase scrapie. Vet. Res. 2010, 6, 49. [Google Scholar] [CrossRef]
- Ma, D.; Li, L. Searching for reliable premortem protein biomarkers for prion diseases: Progress and challenges to date. Expert. Rev. Proteom. 2012, 9, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Chich, J.F.; Schaeffer, B.; Bouin, A.P.; Mouthon, F.; Labas, V.; Larramendy, C.; Deslys, J.P.; Grosclaude, J. Prion infection-impaired functional blocks identified by proteomics enlighten the targets and the curing pathways of an anti-prion drug. Biochim. Biophys. Acta 2007, 1774, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Roostaee, A.R.; Roostaee, M.H.; Roucou, X. An update on prion biology and proteomics. Curr. Proteom. 2010, 7, 36–48. [Google Scholar] [CrossRef]
- Morel, N.; Andreoletti, O.; Grassi, J.; Clement, G. Absolute and relative quantification of sheep brain prion protein (PrP) allelic variants by matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry. Rapid Comm. Mass Spectrom. 2007, 21, 4093–4100. [Google Scholar] [CrossRef]
- Petrakis, S.; Malinowska, A.; Dadlez, M.; Sklaviadis, T. Identification of proteins co-purifying with scrapie infectivity. J. Proteom. 2009, 72, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, A.; Di Francesco, L.; Principe, S.; Mignogna, G.; Sennels, L.; Mancone, C.; Alonzi, T.; Sbriccoli, M.; De Pascalis, A.; Rappsilber, J.; et al. Proteomic profiling of PrP27-30-enriched preparations extracted from the brain of hamsters with experimental scrapie. Proteomics 2009, 9, 3802–3814. [Google Scholar] [CrossRef]
- Kim, B.H.; Jun, Y.C.; Jin, J.K.; Kim, J.I.; Kim, N.H.; Leibold, E.A.; Connor, J.R.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Alteration of iron regulatory proteins (IRP1 and IRP2) and ferritin in the brains of scrapie-infected mice. Neurosci. Lett. 2007, 422, 158–163. [Google Scholar] [CrossRef]
- Dong, C.F.; Shi, S.; Wang, X.F.; An, R.; Li, P.; Chen, J.M.; Wang, X.; Wang, G.R.; Shan, B.; Zhang, B.Y.; et al. The N-terminus of PrP is responsible for interacting with tubulin and fCJD related PrP mutants possess stronger inhibitive effect on microtubule assembly in vitro. Arch. Biochem. Biophys. 2008, 470, 83–92. [Google Scholar] [CrossRef]
- Hur, K.; Kim, J.I.; Choi, S.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. The pathogenic mechanisms of prion diseases. Mech. Ageing Dev. 2002, 123, 1637–1647. [Google Scholar] [CrossRef]
- Kim, N.H.; Park, S.J.; Jin, J.K.; Kwon, M.S.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Increased ferric iron content and iron-induced oxidative stress in the brains of scrapie-infected mice. Brain Res. 2000, 884, 98–103. [Google Scholar] [CrossRef]
- Wisniewski, H.M.; Lossinsky, A.S.; Moretz, R.C.; Vorbrodt, A.W.; Lassmann, H.; Carp, R.I. Increased blood-brain barrier permeability in scrapie-infected mice. J. Neuropathol. Exp. Neurol. 1983, 42, 615–626. [Google Scholar] [CrossRef]
- Kim, J.I.; Choi, S.I.; Kim, N.H.; Jin, J.K.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Oxidative stress and neurodegeneration in prion diseases. Ann. N. Y. Acad. Sci. 2001, 928, 182–186. [Google Scholar] [CrossRef]
- Diedrich, J.F.; Minnigan, H.; Carp, R.I.; Whitaker, J.N.; Race, R.; Frey, W.; Hasse, A.T. Neuropathological changes in scrapie and Alzheimer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J. Virol. 1991, 65, 4759–4768. [Google Scholar] [CrossRef]
- Gao, C.; Lei, Y.J.; Han, J.; Shi, Q.; Chen, L.; Guo, Y.; Gao, Y.J.; Chen, J.M.; Jian, H.Y.; Zhou, W.; et al. Recombinant neural protein PrP can bind with both recombinant and native apolipoprotein E in vitro. Acta Biochim. Biophys. Sin. 2006, 38, 593–601. [Google Scholar] [CrossRef]
- Phuan, P.W.; Zorn, J.A.; Safar, J.; Giles, K.; Prusiner, S.B.; Cohen, F.E.; May, B.C. Discriminating between cellular and misfolded prion protein by using affinity to 9-aminoacridine compounds. J. Gen. Virol. 2007, 88, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Thanomsridetchai, N.; Singhto, N.; Tepsumethanon, V.; Shuangshoti, S.; Wacharapluesadee, S.; Sinchaikul, S.; Chen, S.T.; Hemachudha, T.; Thongboonkerd, V. Comprehensive proteome analysis of hippocampus, brainstem, and spinal cord from paralytic and furious dogs naturally infected with rabies. J. Proteome Res. 2011, 10, 4911–4924. [Google Scholar] [CrossRef]
- Fu, Z.F.; Weihe, E.; Zheng, Y.M.; Schafer, M.K.; Sheng, H.; Corisdeo, S.; Rauscher, F.J., III; Koprowski, H.; Dietzschold, B. Differential effects of rabies and borna disease viruses on immediate-early- and late-response gene expression in brain tissues. J. Virol. 1993, 67, 6674–6681. [Google Scholar] [CrossRef] [PubMed]
- Prosniak, M.; Hooper, D.C.; Dietzschold, B.; Koprowski, H. Effect of rabies virus infection on gene expression in mouse brain. Proc. Natl. Acad. Sci. USA 2001, 98, 2758–2763. [Google Scholar] [CrossRef]
- Dhingra, V.; Li, X.; Liu, Y.; Fu, Z.F. Proteomic profiling reveals that rabies virus infection results in differential expression of host proteins involved in ion homeostasis and synaptic physiology in the central nervous system. J. Neurovirol. 2007, 13, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kasempimolporn, S.; Lumlertdacha, B.; Chulasugandha, P.; Boonchang, S.; Sitprija, V. Alterations in brain cerebral cortex proteome of rabies-infected cat. Southeast Asian J. Trop. Med. Public Health 2014, 45, 808–815. [Google Scholar] [PubMed]
- Mehta, S.M.; Banerjee, S.M.; Chowdhary, A.S. Postgenomics biomarkers for rabies—The next decade of proteomics. Omics J. Integrat. Biol. 2015, 19, 67–79. [Google Scholar] [CrossRef]
- Mehta, S.; Sreenivasamurthy, S.; Banerjee, S.; Mukherjee, S.; Prasad, K.; Chowdhary, A. Pathway analysis of proteomics profiles in rabies infection: Towards future biomarkers? Omics J. Intergrativ. Biol. 2016, 20, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.; Rosa, N.; Esteves, E.; Correia, M.J.; Arrais, J.; Ribeiro, P.; Vala, H.; Barros, M. CanisOme—The protein signatures of Canis lupus familiaris diseases. J. Proteom. 2016, 136, 193–201. [Google Scholar] [CrossRef]
- Venugopal, A.K.; Ghantasala, S.S.K.; Selvan, L.D.N.; Mahadevan, A.; Renuse, S.; Kumar, P.; Pawar, H.; Sahasrabhuddhe, N.A.; Suja, M.S.; Ramachandra, Y.L.; et al. Quantitative proteomics for identifying biomarkers for rabies. Clin. Proteom. 2013, 10, 3. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Wang, Q.; Jung, K.; Langel, S.N.; Malik, Y.S.; Saif, L.J. Porcine coronaviruses. In Emerging and Transboundary Animal Viruses; Malik, Y., Singh, R., Yadav, M., Eds.; Springer: Singapore, 2020; pp. 79–110. [Google Scholar]
- Mora-Diaz, J.C.; Pineyro, P.E.; Houston, E.; Zimmerman, J.; Gimenez-Lirola, L.G. Porcine hemagglutinating encephalomyelitis virus: A review. Front. Vet. Sci. 2019, 6, 53. [Google Scholar] [CrossRef]
- Song, D.; Moon, H.; Kang, B. Porcine epidemic diarrhea: A review of current epidemiology and available vaccines. Clin. Exp. Vaccine Res. 2015, 4, 166–176. [Google Scholar] [CrossRef]
- Licitra, B.N.; Duhamel, G.E.; Whittaker, G.R. Canine enteric coronaviruses: Emerging viral pathogens with distinct recombinant spike proteins. Viruses 2014, 6, 3363–3376. [Google Scholar] [CrossRef]
- Erles, K.; Brownlie, J. Canine respiratory coronavirus: An emerging pathogen in the canine infectious respiratory disease complex. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 815–825. [Google Scholar] [CrossRef]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Wang, N.; Li, S.Y.; Yang, X.L.; Huang, H.M.; Zhang, Y.J.; Guo, H.; Luo, C.M.; Miller, M.; Zhu, G.; Chmura, A.A.; et al. Serological evidence of bat SARS-related coronavirus infection in humans, China. Virol. Sin. 2018, 33, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Grenga, L.; Armengaud, J. Proteomics in the COVID-19 battlefield: First semester check-up. Proteomics 2021, 21, e2000198. [Google Scholar] [CrossRef]
- Giri, R.; Bhardwaj, T.; Shegane, M.; Gehi, B.R.; Kumar, P.; Gadhave, K.; Oldfield, C.J.; Uversky, V.N. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell. Mol. Life Sci. 2021, 78, 1655–1688. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhao, S.; Teng, T.; Abdalla, A.E.; Zhu, W.; Xie, L.; Wang, Y.; Guo, X. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 2020, 12, 244. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.Q.; Oubraham, L.; Wang, A.Q.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.S.; Kreidl, M.; et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020, 11, 11–18. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Thomas, T.; Dzieciatkowska, M.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hod, E.A.; Spitalnik, S.L.; Hansen, K.C. Serum proteomics in COVID-19 patients: Altered coagulation and complement status as a function of IL-6 level. J. Proteom. Res. 2020, 19, 4417–4427. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–462. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Kramer, L.D.; Li, J.; Shi, P.Y. West Nile virus. Lancet Neurol. 2007, 6, 171–181. [Google Scholar] [CrossRef]
- Valiakos, G.; Athanasiou, L.V.; Touloudi, A.; Papatsiros, V.; Spyrou, V.; Petrovska, L.; Billinis, C. West Nile Virus: Basic Principles, Replication Mechanism, Immune Response and Important Genetic Determinants of Virulence. In Viral Replication; Rosas-Acosta, G., Ed.; InTech Open: London, UK, 2013; pp. 43–68. [Google Scholar]
- Altamura, L.A.; Cazares, L.H.; Coyne, S.R.; Jaissle, J.G.; Jespersen, A.M.; Ahmed, S.; Wasieloski, L.P.; Garrison, J.; Kulesh, D.A.; Brueggemann, E.E.; et al. Inactivation of West Nile virus in serum with heat, ionic detergent, and reducing agent for proteomic applications. J. Virol. Methods 2017, 248, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, J.; Ye, J.; Ashraf, U.; Chen, Z.; Zhu, B.; He, W.; Xu, Q.; Wei, Y.; Chen, H.; et al. Quantitative label-free phosphoproteomics reveals differentially regulated protein phosphorylation involved in West Nile Virus-induced host inflammatory response. J. Proteome Res. 2015, 14, 5157–5168. [Google Scholar] [CrossRef]
- Tognotti, E. Influenza pandemics: A historical retrospect. J. Infect. Dev. Ctries. 2009, 3, 331–334. [Google Scholar] [CrossRef]
- Ma, W.; Kahn, R.E.; Richt, J.A. The pig as a mixing vessel for influenza viruses: Human and veterinary implications. J. Mol. Genet. Med. 2008, 3, 158–166. [Google Scholar] [CrossRef]
- Zhu, J.; Zou, W.; Jia, G.; Zhou, H.; Hu, Y.; Peng, M.; Chen, H.; Jin, M. Analysis of cellular proteome alterations in porcine alveolar macrophage cells infected with 2009 (H1N1) and classical swine H1N1 influenza viruses. J. Proteom. 2012, 75, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Dove, B.K.; Surtees, R.; Bean, T.J.; Munday, D.; Wise, H.M.; Digard, P.; Carroll, M.W.; Ajuh, P.; Barr, J.N.; Hiscox, J.A. A quantitative proteomic analysis of lung epithelial (A549) cells infected with 2009 pandemic influenza A virus using stable isotope labelling with amino acids in cell culture. Proteomics 2012, 12, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Abd Raman, H.S.; Tan, S.; August, J.T.; Khan, A.M. Dynamics of Influenza A (H5N1) virus protein sequence diversity. Peer J. 2020, 7, e7954. [Google Scholar] [CrossRef]
- Sun, J.F.; Han, Z.X.; Shao, Y.H.; Cao, Z.Z.; Kong, X.G.; Liu, S.W. Comparative proteome analysis of tracheal tissues in response to infectious bronchitis coronavirus, Newcastle disease virus, and avian influenza virus H9 subtype virus infection. Proteomics 2014, 14, 1403–1423. [Google Scholar] [CrossRef]
- Zou, W.; Ke, J.; Zhang, A.; Zhou, M.; Liao, Y.; Zhu, J.; Zhou, H.; Tu, J.; Chen, H.; Jin, M. Proteomics analysis of differential expression of chicken brain tissue proteins in response to the neurovirulent H5N1 avian influenza virus infection. J. Proteome Res. 2010, 9, 3789–3798. [Google Scholar] [CrossRef]
- Sun, N.; Sun, W.; Li, S.; Yang, J.; Yang, L.; Quan, G.; Gao, X.; Wang, Z.; Cheng, X.; Li, Z.; et al. Proteomics analysis of cellular proteins co-immunoprecipitated with nucleoprotein of influenza A virus (H7N9). Int. J. Mol. Sci. 2015, 16, 25982–25998. [Google Scholar] [CrossRef]
- Sui, Z.; Wen, B.; Gao, Z.; Chen, Q. Fusion-related host proteins are actively regulated by NA during influenza infection as revealed by quantitative proteomics analysis. PLoS ONE 2014, 9, e105947. [Google Scholar]
- Wu, H.; Zhang, S.; Huo, C.; Zou, S.; Lian, Z.; Hu, Y. iTRAQ-based proteomic and bioinformatic characterization of human mast cells upon infection by the influenza A virus strains H1N1 and H5N1. FEBS Lett. 2019, 593, 2612–2627. [Google Scholar] [CrossRef]
- Mitchell, H.D.; Eisfeld, A.J.; Stratton, K.G.; Heller, N.C.; Bremer, L.M.; Wen, J.; McDermott, J.E.; Gralinski, L.E.; Sims, A.C.; Le, M.Q.; et al. The role of EGFR in influenza pathogenicity: Multiple network-based approaches to identify a key regulator of non-lethal infections. Front. Cell Dev. Biol. 2019, 7, 200. [Google Scholar] [CrossRef]
- Sender, V.; Hentrich, K.; Pathak, A.; Ler, A.T.Q.; Embaie, B.T.; Lundstrom, S.L.; Gaetani, M.; Bergstrand, J.; Nakamoto, R.; Sham, L.T.; et al. Capillary leakage provides nutrients and antioxidants for rapid pneumococcal proliferation in influenza-infected lower airways. Proc. Natl. Acad. Sci. USA 2020, 117, 31386–31397. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, Q.; Zhao, W.; Sun, W.; Yang, J.; Liu, N. Proteomics in influenza research: The emerging role of post-translational modifications. J. Proteom. Res. 2021, 20, 110–121. [Google Scholar] [CrossRef]
- Weber, A.; Dam, S.; Saul, V.V.; Kuznetsova, I.; Müller, C.; Fritz-Wolf, K.; Becker, K.; Linne, U.; Gu, H.; Stokes, M.P.; et al. Phosphoproteome analysis of cells infected with adapted and non-adapted Influenza A virus reveals novel pro- and antiviral signaling networks. J. Virol. 2019, 93, e00528-19. [Google Scholar] [CrossRef] [PubMed]
- Hawksworth, A.; Jayachander, M.; Hester, S.; Mohammed, S.; Hutchinson, E. Proteomics as a tool for live attenuated influenza vaccine characterisation. Vaccine 2020, 38, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bannantine, J.P.; Campo, J.J.; Randall, A.; Grohn, Y.T.; Katani, R.; Schilling, M.; Radzio-Basu, J.; Kapur, V. Identification of sero-reactive antigens for the early diagnosis of Johne’s disease in cattle. PLoS ONE 2017, 12, e0184373. [Google Scholar] [CrossRef] [PubMed]
- Hughes, V.; Smith, S.; Garcia-Sanchez, A.; Sales, J.; Stevenson, K. Proteomic comparison of Mycobacterium avium subspecies paratuberculosis grown in vitro and isolated from clinical cases of ovine paratuberculosis. Microbiology 2007, 153, 196–205. [Google Scholar] [CrossRef][Green Version]
- Hughes, V.; Bannantine, J.P.; Denham, S.; Smith, S.; Garcia-Sanchez, A.; Sales, J.; Paustian, M.L.; Mclean, K.; Stevenson, K. Immunogenicity of proteome-determined Mycobacterium avium subsp. paratuberculosis-specific proteins in sheep with paratuberculosis. Clin. Vaccine Immunol. 2008, 15, 1824–1833. [Google Scholar] [PubMed]
- Hughes, V.; Garcia-Sanchez, A.; Smith, S.; Mclean, K.; Lainson, A.; Nath, M.; Stevenson, K. Proteome-determined type-specific proteins of Mycobacterium avium subspecies paratuberculosis. Vet. Microbiol. 2012, 158, 153–162. [Google Scholar] [CrossRef]
- Hughes, V.; Denham, S.; Bannantine, J.P.; Chianni, F.; Kerr, K.; May, L.; McLuckie, J.; Nath, M.; Stevenson, K. Interferon gamma responses to proteome-determined specific recombinant proteins: Potential as diagnostic markers for ovine Johne’s disease. Vet. Immunol. Immunopathol. 2013, 155, 197–204. [Google Scholar] [CrossRef]
- Zhong, L.; Taylor, D.; Begg, D.J.; Whittington, R.J. Biomarker discovery for ovine paratuberculosis (Johne’s disease) by proteomic serum profiling. Comp. Immunol. Microbiol. Infect. Dis. 2011, 34, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Roupie, V.; Noël-Georis, I.; Rosseels, V.; Walravens, K.; Govaerts, M.; Huygen, K.; Wattiez, R. Antigen discovery: A postgenomic approach to paratuberculosis diagnosis. Proteomics 2007, 7, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, V.; Villar, M.; Martín-Hernando, M.P.; Vidal, D.; Höfle, U.; Gortazar, C.; Kocan, K.M.; Vázquez, J.; de la Fuente, J. Proteomic and transcriptomic analyses of differential stress/inflammatory responses in mandibular lymph nodes and oropharyngeal tonsils of European wild boars naturally infected with Mycobacterium bovis. Proteomics 2007, 7, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Seth, M.; Lamont, E.A.; Janagama, H.K.; Widdel, A.; Vulchanova, L.; Stabel, J.R.; Waters, W.R.; Palmer, M.V.; Sreevatsan, S. Biomarker discovery in subclinical mycobacterial infections of cattle. PLoS ONE 2009, 4, e5478. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Krutzik, S.R.; Modlin, R.L. Therapeutic implications of the TLR and VDR partnership. Trends Mol. Med. 2007, 13, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Lamont, E.A.; Janagama, H.K.; Ribeiro-Lima, J.; Vulchanova, L.; Seth, M.; Yang, M.; Kurmi, K.; Waters, W.R.; Thacker, T.; Sreevatsan, S. Circulating Mycobacterium bovis peptides and host response proteins as biomarkers for unambiguous detection of subclinical infection. J. Clin. Microbiol. 2014, 52, 536–543. [Google Scholar] [CrossRef]
- Li, P.; Wang, R.; Dong, W.; Hu, L.; Zong, B.; Zhang, Y.; Wang, X.; Guo, A.; Zhang, A.; Xiang, Y.; et al. Comparative proteomics analysis of human macrophages infected with virulent Mycobacterium bovis. Front. Cell. Infect. Microbiol. 2017, 7, 65. [Google Scholar] [CrossRef]
- Lopez, V.; van der Heijden, E.; Villar, M.; Michel, A.; Alberdi, P.; Gortázar, C.; Rutten, V.; de la Fuente, J. Comparative proteomics identified immune response proteins involved in response to vaccination with heat-inactivated Mycobacterium bovis and mycobacterial challenge in cattle. Vet. Immunol. Immunopathol. 2018, 206, 54–64. [Google Scholar] [CrossRef]
- Allerberger, F.; Wagner, M. Listeriosis: A resurgent foodborne infection. Clin. Microbiol. Infect. 2010, 16, 16–23. [Google Scholar] [CrossRef]
- Cacace, G.; Mazzeo, M.F.; Sorrentino, A.; Spada, V.; Malorni, A.; Siciliano, R.A. Proteomics for the elucidation of cold adaptation mechanisms in Listeria monocytogenes. J. Proteom. 2010, 73, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Schmidt, T.B.; Nanduri, B.; Pendarvis, K.; Pittman, J.R.; Thornton, J.A.; Grissett, J.; Donaldson, J.R. Proteomic analysis of the response of Listeria monocytogenes to bile salts under anaerobic conditions. J. Med. Microbiol. 2013, 62, 25–35. [Google Scholar] [CrossRef]
- Jadhav, S.; Sevior, D.; Bhave, M.; Palombo, E.A. Detection of Listeria monocytogenes from selective enrichment broth using MALDI-TOF mass spectrometry. J. Proteom. 2014, 97, 100–106. [Google Scholar] [CrossRef]
- Yuan, C.L.; Zhu, C.X.; Hua, X.G. Bartonella henselae infection and its effects on human health. Rev. Med. Microbiol. 2011, 22, 67–72. [Google Scholar]
- Yuan, C.L.; Zhu, C.X.; Wu, Y.B.; Pan, X.Y.; Hua, X.G. Bacteriological and molecular identification of Bartonella species in cats from different regions of China. PLoS Negl. Trop. Dis. 2011, 5, e1301. [Google Scholar] [CrossRef] [PubMed]
- Chomel, B.B.; Boulouis, H.J.; Maruyama, S.; Breitschwerdt, E.B. Bartonella spp. in pets and effect on human health. Emerg. Infect. Dis. 2006, 12, 389–394. [Google Scholar] [CrossRef]
- Young, D.B. Chaperonins and the immune response. Semin. Cell Biol. 1990, 1, 27–35. [Google Scholar] [PubMed]
- Haake, D.A.; Summers, T.A.; McCoy, A.M.; Schwartzman, W. Heat shock response and groEL sequence of Bartonella henselae and Bartonella quintana. Microbiology 1997, 143, 2807–2815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eberhardt, C.; Engelmann, S.; Kusch, H.; Albrecht, D.; Hecker, M.; Autenrieth, I.B.; Kempf, V.A. Proteomic analysis of the bacterial pathogen Bartonella henselae and identification of immunogenic proteins for serodiagnosis. Proteomics 2009, 9, 1967–1981. [Google Scholar] [CrossRef]
- Blasco, J.M.; Molina-Flores, B. Control and eradication of Brucella melitensis infection in sheep and goats. Vet. Clin. N. Am. Food Anim. Pract. 2011, 27, 95–104. [Google Scholar] [CrossRef]
- Wagner, M.A.; Eschenbrenner, M.; Horn, T.A.; Kraycer, J.A.; Mujer, C.V.; Hagius, S.; Elzer, P.; DelVecchio, V.G. Global analysis of the Brucella melitensis proteome: Identification of proteins expressed in laboratory-grown culture. Proteomics 2002, 2, 1047–1060. [Google Scholar] [CrossRef]
- Mavromati, J. Brucellosis and proteomics: An approach in Albania. In Farm Animal Proteomics 2012, Proceedings of the 3rd Managing Committee Meeting and 2nd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Algarve, Portugal, 27 March 2012; Rodrigues, P., Eckersall, D., de Almeida, A., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2012; pp. 80–83. [Google Scholar]
- Garcia-Bates, T.M.; Baglole, C.J.; Bernard, M.P.; Murant, T.I.; Simpson-Haidaris, P.J.; Phipps, R.P. Peroxisome proliferator-activated receptor gamma ligands enhance human B cell antibody production and differentiation. J. Immunol. 2009, 183, 6903–6912. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Melzer, F.; Weise, C.; Neubauer, H.; Roesler, U.; Murugaiyan, J. Proteomics-based identification of immunodominant proteins of Brucellae using sera from infected hosts points towards enhanced pathogen survival during the infection. Biochem. Biophys. Res. Commun. 2015, 456, 202–206. [Google Scholar] [CrossRef]
- Barbosa Pauletti, R.; Reinato Stynen, A.P.; Pinto da Silva Mol, J.; Seles Dorneles, E.M.; Alves, T.M.; de Sousa Moura Souto, M.; Minharro, S.; Heinemann, M.B.; Lage, A.P. Reduced susceptibility to rifampicin and resistance to Multiple Antimicrobial Agents among Brucella abortus Isolates from Cattle in Brazil. PLoS ONE 2015, 10, e0132532. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.; Marchesini, M.I. Proteomics of Brucella. Proteomes 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Hayat, Z.; Khan, H.; Ahmad, I.; Habib, H.; Hayat, K. Antibiotics in the management of brucellosis. Gomal J. Med. Sci. 2018, 16, 114–116. [Google Scholar]
- Eschenbrenner, M.; Horn, T.A.; Wagner, M.A.; Mujer, C.V.; Miller-Scandle, T.L.; DelVecchio, V.G. Comparative proteome analysis of laboratory grown Brucella abortus 2308 and Brucella melitensis 16M. J. Proteome Res. 2006, 5, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Eravci, M.; Weise, C.; Roesler, U.; Melzer, F.; Sprague, L.D.; Neubauer, H.; Murugaiyan, J. Comprehensive identification of immunodominant proteins of Brucella abortus and Brucella melitensis using antibodies in the sera from naturally infected hosts. Int. J. Mol. Sci. 2016, 17, 659. [Google Scholar] [CrossRef]
- Connolly, J.P.; Comerci, D.; Alefantis, T.G.; Walz, A.; Quan, M.; Chafin, R.; Grewal, P.; Mujer, C.V.; Ugalde, R.A.; DelVecchio, V.G. Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics 2006, 6, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- Pajuaba, A.C.; Silva, D.A.; Almeida, K.C.; Cunha-Junior, J.P.; Pirovani, C.P.; Camillo, L.R.; Mineo, J.R. Immunoproteomics of Brucella abortus reveals differential antibody profiles between S19-vaccinated and naturally infected cattle. Proteomics 2012, 12, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Khan, M.T.; Iqbal, A. Identification of novel drug targets for humans and potential vaccine targets for cattle by subtractive genomic analysis of Brucella abortus strain 2308. Microb. Pathogen. 2019, 137, 103731. [Google Scholar] [CrossRef] [PubMed]
- Dohre, S.K.; Kamthan, A.; Singh, S.; Alam, S.I.; Kumar, S. Identification of a new diagnostic antigen for glanders using immunoproteome analysis. Comp. Immunol. Microbiol. Infect. Dis. 2017, 53, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, K.; Meyers, J.L.; Deshazer, D.; Riggins, R.N.; Halasohoris, S.; England, M.; Ribot, W.; Norris, S.L.; Waag, D.M. Detection of the host immune response to Burkholderia mallei heat-shock proteins GroEL and DnaK in a glanders patient and infected mice. Diagn. Microbiol. Infect. Dis. 2007, 59, 137–147. [Google Scholar] [CrossRef]
- Yazdansetad, S.; Mosavari, N.; Tadayon, K.; Mehregan, I. Development of an immunoblotting assay for serodiagnosis of Burkholderia mallei infection: The whole-cell proteome-based paradigm. Iran J. Microbiol. 2019, 11, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Seal, B.S.; Hiett, K.L.; Kuntz, R.L.; Woolsey, R.; Schegg, K.M.; Ard, M.; Stintzi, A. Proteomic analyses of a robust versus a poor chicken gastrointestinal colonizing isolate of Campylobacter jejuni. J. Proteome Res. 2007, 6, 4582–4591. [Google Scholar] [CrossRef]
- Wu, Z.; Sahin, O.; Wang, F.; Zhang, Q. Proteomic identification of immunodominant membrane-related antigens in Campylobacter jejuni associated with sheep abortion. J. Proteom. 2014, 99, 111–122. [Google Scholar] [CrossRef]
- Moser, I.; Schroeder, W.; Salnikow, J. Campylobacter jejuni major outer membrane protein and a 59-kDa protein are involved in binding to fibronectin and INT 407 cell membranes. FEMS Microbiol. Lett. 1997, 157, 233–238. [Google Scholar] [CrossRef]
- Islam, A.; Raghupathy, R.; Albert, M.J. Recombinant PorA, the major outer membrane protein of Campylobacter jejuni, provides heterologous protection in an adult mouse intestinal colonization model. Clin. Vaccine Immunol. 2010, 17, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.; Phansopa, C.; Suwannachart, C.; Craven, C.J.; Rafferty, J.B.; Kelly, D.J. The virulence factor PEB4 (Cj0596) and the periplasmic protein Cj1289 are two structurally related SurA-like chaperones in the human pathogen Campylobacter jejuni. J. Biol. Chem. 2011, 286, 21254–21265. [Google Scholar] [CrossRef]
- Asakura, H.; Kawamoto, K.; Murakami, S.; Tachibana, M.; Kurazono, H.; Makino, S.; Yamamoto, S.; Igimi, S. Ex vivo proteomics of Campylobacter jejuni 81-176 reveal that FabG affects fatty acid composition to alter bacterial growth fitness in the chicken gut. Res. Microbiol. 2016, 167, 63–71. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.L.; Burchmore, R.J.; Sparks, N.H.; Eckersall, P.D. The effect of microbial challenge on the intestinal proteome of broiler chickens. Proteome Sci. 2017, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, E.; Williams, P.; Ray, W. Proteomic evaluation of chicken brush-border membrane during the early posthatch period. J. Proteom. Res. 2010, 9, 4628–4639. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Gu, X.; Wang, X. Overexpression of heat shock protein 70 and its relationship to intestine under acute heat stress in broilers: 1. Intestinal structure and digestive function. Poult. Sci. 2012, 91, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-T.; Li, Y.-H.; Chou, I.-P.; Hsieh, Y.-H.; Chen, B.-J.; Chen, C.-Y. Albusin B modulates lipid metabolism and increases antioxidant defense in broiler chickens by a proteomic approach. J. Sci. Food Agric. 2013, 93, 284–292. [Google Scholar] [CrossRef]
- Zhang, J.; Li, C.; Tang, X.; Lu, Q.; Sa, R.; Zhang, H. Proteome changes in the small intestinal mucosa of broilers (Gallus gallus) induced by high concentrations of atmospheric ammonia. Proteome Sci. 2015, 13, 9. [Google Scholar] [CrossRef]
- Ayllón, N.; Jiménez-Marín, Á.; Argüello, H.; Zaldívar-López, S.; Villar, M.; Aguilar, C.; Moreno, A.; De La Fuente, J.; Garrido, J.J. Comparative proteomics reveals differences in host-pathogen interaction between infectious and commensal relationship with Campylobacter jejuni. Front. Cell Infect. Microbiol. 2017, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ohki, M.; Urata, A.; Ohshiro, S.; Tarigan, E.; Kiatsomphob, S.; Vetchapitak, T.; Sato, H.; Misawa, N. Detection and identification of adhesins involved in adhesion of Campylobacter jejuni to chicken skin. Int. J. Food Microbiol. 2021, 337, 108929. [Google Scholar] [CrossRef] [PubMed]
- Arricau-Bouvery, N.; Souriau, A.; Lechopier, P.; Rodolakis, A. Excretion of Coxiella burnetii during an experimental infection of pregnant goats with an abortive goat strain CbC1. Ann. N. Y. Acad. Sci. 2003, 990, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Rodolakis, A.; Berri, M.; Héchard, C.; Caudron, C.; Souriau, A.; Bodier, C.C.; Blanchard, B.; Camuset, P.; Devillechaise, P.; Natorp, J.C.; et al. Comparison of Coxiella burnetii shedding in milk of dairy bovine, caprine, and ovine herds. J. Dairy Sci. 2007, 90, 5352–5360. [Google Scholar] [CrossRef]
- Van den Brom, R.; de Jong, A.; van Engelen, E.; Heuvelink, A.; Vellema, P. Zoonotic risks of pathogens from sheep and their milk borne transmission. Small Rumin. Res. 2020, 189, 106123. [Google Scholar] [CrossRef]
- De Bruin, A.; van der Plaats, R.Q.; de Heer, L.; Paauwe, R.; Schimmer, B.; Vellema, P.; van Rotterdam, B.J.; van Duynhoven, Y.T. Detection of Coxiella burnetii DNA on small-ruminant farms during a Q fever outbreak in the Netherlands. Appl. Environ. Microbiol. 2012, 78, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Vellema, P.; van den Brom, R. The rise and control of the 2007-2012 human Q fever outbreaks in the Netherlands. Small Rumin. Res. 2014, 118, 69–78. [Google Scholar] [CrossRef]
- Papadioti, A.; Markoutsa, S.; Vranakis, I.; Tselentis, Y.; Karas, M.; Psaroulaki, A.; Tsiotis, G.A. Proteomic approach to investigate the differential antigenic profile of two Coxiella burnetii strains. J. Proteom. 2011, 74, 1150–1159. [Google Scholar] [CrossRef]
- Mucha, R.; Bencurova, E.; Cepkova, M.; Mlynarcik, P.; Madar, M.; Pulzova, L.; Hresko, S.; Bhide, M. Adhesion of Francisella to endothelial cells is also mediated by OmpA: ICAM-1 interaction. In Farm Animal Proteomics 2012, Proceedings of the 3rd Managing Committee Meeting and 2nd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Algarve, Portugal, 27 March 2012; Rodrigues, P., Eckersall, D., de Almeida, A., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2012; pp. 94–97. [Google Scholar]
- Gaur, R.; Alam, S.I.; Kamboj, D.V. Immunoproteomic analysis of antibody response of rabbit host against heat-killed Francisella tularensis live vaccine strain. Curr. Microbiol. 2017, 74, 499–507. [Google Scholar] [CrossRef]
- Evangelopoulou, G.; Filioussis, G.; Kritas, S.; Christodoulopoulos, G.; Triantafillou, E.A.; Burriel, A.R. Colonisation of pig gallbladders with Salmonella species important to public health. Vet. Rec. 2015, 176, 174. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Park, S.; Barate, A.K.; Truong, Q.L.; Han, J.H.; Jung, C.H.; Yoon, J.W.; Cho, S.; Hahn, T.W. Proteomic analysis of outer membrane proteins in Salmonella enterica Enteritidis. J. Microbiol. Biotechnol. 2015, 25, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Arce, C.; Lucena, C.; Moreno, A.; Garrido, J.J. Proteomic analysis of intestinal mucosa responses to Salmonella enterica serovar Typhimurium in naturally infected pig. Comp. Immunol. Microbiol. Infect. Dis. 2014, 37, 59–67. [Google Scholar] [CrossRef]
- Martins, R.P.; Collado-Romero, M.; Martínez-Gomáriz, M.; Carvajal, A.; Gil, C.; Lucena, C.; Moreno, A.; Garrido, J.J. Proteomic analysis of porcine mesenteric lymph-nodes after Salmonella typhimurium infection. J. Proteom. 2012, 75, 4457–4470. [Google Scholar] [CrossRef]
- Collado-Romero, M.; Martins, R.P.; Arce, C.; Moreno, Á.; Lucena, C.; Carvajal, A.; Garrido, J.J. An in vivo proteomic study of the interaction between Salmonella Typhimurium and porcine ileum mucosa. J. Proteom. 2012, 75, 2015–2026. [Google Scholar] [CrossRef]
- Collado-Romero, M.; Aguilar, C.; Arce, C.; Lucena, C.; Codrea, M.C.; Morera, L.; Bendixen, E.; Moreno, Á.; Garrido, J.J. Quantitative proteomics and bioinformatic analysis provide new insight into the dynamic response of porcine intestine to Salmonella Typhimurium. Front. Cell. Infect. Microbiol. 2015, 5, 64. [Google Scholar] [CrossRef]
- Revajova, V.; Levkut, M.; Laukova, A.; Herich, R.; Sevcikova, Z.; Kolesarova, M. Immunoreactivity to Salmonella infection in chicks protected with Enteroccocus administration. Immunology 2012, 137, 628. [Google Scholar]
- Smirnov, A.; Perez, R.; Amit-Romach, E.; Sklan, D.; Uni, Z. Mucin dynamics andmicrobial populations in chicken small intestine are changed by dietaryprobiotic and antibiotic growth promoter supplementation. J. Nutr. 2005, 135, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Levkut, M.; Revajova, V.; Laukova, A.; Sevcikova, Z.; Spisakova, V.; Faixova, Z.; Levkutova, M.; Strompfova, V.; Pistl, J.; Levkut, M. Leukocytic responses and intestinal mucin dynamics of broilers protected with Enterococcus faecium EF55 and challenged with Salmonella Enteritidis. Res. Vet. Sci. 2012, 93, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Polansky, O.; Seidlerova, Z.; Faldynova, M.; Sisak, F.; Rychlik, I. Protein expression in the liver and blood serum in chickens in response to Salmonella Enteritidis infection. Vet. Immunol. Immunopathol. 2018, 205, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Sekelova, Z.; Polansky, O.; Stepanova, H.; Fedr, R.; Faldynova, M.; Rychlik, I.; Vlasatikova, L. Different roles of CD4, CD8 and γδ T-lymphocytes in naive and vaccinated chickens during Salmonella enteritidis infection. Proteomics 2017, 17, 1700073. [Google Scholar] [CrossRef] [PubMed]
- Talagrand-Reboul, E.; Westermann, B.; Raess, M.A.; Schnell, G.; Cantero, P.; Barthel, C.; Ehret-Sabatier, L.; Jaulhac, B.; Boulanger, N. Proteomic as an exploratory approach to develop vaccines against tick-borne diseases using Lyme borreliosis as a test case. Vaccines 2020, 8, 463. [Google Scholar] [CrossRef]
- Bhide, M.; Bhide, K.; Pulzova, L.; Madar, M.; Mlynarcik, P.; Bencurova, E.; Hresko, S.; Mucha, R. Variable regions in the sushi domains 6-7 and 19-20 of factor H in animals and human lead to change in the affinity to factor H binding protein of Borrelia. J. Proteom. 2012, 75, 4520–4528. [Google Scholar] [CrossRef]
- Bencurova, E.; Gupta, S.K.; Oskoueian, E.; Bhide, M.; Dandekar, T. Omics and bioinformatics applied to vaccine development against Borrelia. Mol. Omics 2018, 14, 330–340. [Google Scholar] [CrossRef]
- Ting, T.X.; Amran, F.B.; Ahmad, N.; Abdul-Rahman, P.S. Integration of omics research in discovery of biomarkers for leptospirosis diagnosis and vaccine development. Southeast Asian J. Trop. Med. Public Health 2019, 50, 300–324. [Google Scholar]
- Humphryes, P.C.; Weeks, M.E.; Coldham, N.G. Characterisation of the proteome of Leptospira interrogans Serovar Canicola as a resource for the identification of common serovar immunogenic proteins. Int. J. Proteom. 2014, 2014, 572901. [Google Scholar] [CrossRef]
- Eshghi, A.; Cullen, P.A.; Cowen, L.; Zuerner, R.L.; Cameron, C.E. Global proteome analysis of Leptospira interrogans. J. Proteome Res. 2009, 8, 4564–4578. [Google Scholar] [CrossRef] [PubMed]
- Malmström, J.; Beck, M.; Schmidt, A.; Lange, V.; Deutsch, E.W.; Aebersold, R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature 2009, 460, 762–765. [Google Scholar] [CrossRef]
- Vieira, M.L.; Pimenta, D.C.; de Morais, Z.M.; Vasconcellos, S.A.; Nascimento, A.L. Proteome analysis of Leptospira interrogans virulent strain. Open Microbiol. J. 2009, 3, 69–74. [Google Scholar] [CrossRef]
- Cao, X.J.; Dai, J.; Xu, H.; Nie, S.; Chang, X.; Hu, B.Y.; Sheng, Q.H.; Wang, L.S.; Ning, Z.B.; Li, Y.X.; et al. High-coverage proteome analysis reveals the first insight of protein modification systems in the pathogenic spirochete Leptospira interrogans. Cell Res. 2010, 20, 197–210. [Google Scholar] [CrossRef]
- Zhong, Y.; Chang, X.; Cao, X.J.; Zhang, Y.; Zheng, H.; Zhu, Y.; Cai, C.; Cui, Z.; Zhang, Y.; Li, Y.Y.; et al. Comparative proteogenomic analysis of the Leptospira interrogans virulence-attenuated strain IPAV against the pathogenic strain 56601. Cell Res. 2011, 21, 1210–1229. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, Y.; Zhu, Y.; Yin, H.; Zhuang, X.; Zhu, W.; Guo, X.; Qin, J. Extracellular proteome analysis of Leptospira interrogans serovar Lai. Omics 2013, 17, 527–535. [Google Scholar] [CrossRef]
- Thongboonkerd, V.; Chiangjong, W.; Saetun, P.; Sinchaikul, S.; Chen, S.T.; Kositanont, U. Analysis of differential proteomes in pathogenic and non-pathogenic Leptospira: Potential pathogenic and virulence factors. Proteomics 2009, 9, 3522–3534. [Google Scholar] [CrossRef]
- Cullen, P.A.; Cordwell, S.J.; Bulach, D.M.; Haake, D.A.; Adler, B. Global analysis of outer membrane proteins from Leptospira interrogans serovar Lai. Infect. Immun. 2002, 70, 2311–2318. [Google Scholar] [CrossRef]
- Haake, D.A.; Matsunaga, J. Characterization of the leptospiral outer membrane and description of three novel leptospiral membrane proteins. Infect. Immun. 2002, 70, 4936–4945. [Google Scholar] [CrossRef]
- Nally, J.E.; Whitelegge, J.P.; Aguilera, R.; Pereira, M.M.; Blanco, D.R.; Lovett, M.A. Purification and proteomic analysis of outer membrane vesicles from a clinical isolate of Leptospira interrogans serovar Copenhageni. Proteomics 2005, 5, 144–152. [Google Scholar] [CrossRef]
- Nally, J.E.; Whitelegge, J.P.; Bassilian, S.; Blanco, D.R.; Lovett, M.A. Characterization of the outer membrane proteome of Leptospira interrogans expressed during acute lethal infection. Infect. Immun. 2007, 75, 766–773. [Google Scholar] [CrossRef]
- Monahan, A.M.; Callanan, J.J.; Nally, J.E. Proteomic analysis of Leptospira interrogans shed in urine of chronically infected hosts. Infect. Immun. 2008, 76, 4952–4958. [Google Scholar] [CrossRef] [PubMed]
- Nally, J.E.; Monahan, A.M.; Miller, I.S.; Bonilla-Santiago, R.; Souda, P.; Whitelegge, J.P. Comparative proteomic analysis of differentially expressed proteins in the urine of reservoir hosts of leptospirosis. PLoS ONE 2011, 6, e26046. [Google Scholar] [CrossRef]
- Srikram, A.; Zhang, K.; Bartpho, T.; Lo, M.; Hoke, D.E.; Sermswan, R.W.; Adler, B.; Murray, G.L. Cross-protective immunity against leptospirosis elicited by a live, attenuated lipopolysaccharide mutant. J. Infect. Dis. 2011, 203, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.P.; Choudhury, B.; Matthias, M.M.; Baga, S.; Bandyopadhya, K.; Vinetz, J.M. Comparative analysis of lipopolysaccharides of pathogenic and intermediately pathogenic Leptospira species. BMC Microbiol. 2015, 15, 244. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.; de Morais, Z.M.; Gonçales, A.P.; Romero, E.C.; Vasconcellos, S.A.; Nascimento, A.L. Characterization of novel OmpA-like protein of Leptospira interrogans that binds extracellular matrix molecules and plasminogen. PLoS ONE 2011, 6, e21962. [Google Scholar] [CrossRef]
- Cullen, P.A.; Xu, X.; Matsunaga, J.; Sanchez, Y.; Ko, A.I.; Haake, D.A.; Adler, B. Surfaceome of Leptospira spp. Infect. Immun. 2005, 73, 4853–4863. [Google Scholar] [CrossRef] [PubMed]
- Le Maréchal, C.; Jan, G.; Even, S.; McCulloch, J.A.; Azevedo, V.; Thiéry, R.; Vautor, E.; Le Loir, Y. Development of serological proteome analysis of mastitis by Staphylococcus aureus in ewes. J. Microbiol. Methods 2009, 79, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Mansor, R.; Mullen, W.; Albalat, A.; Zerefos, P.; Mischak, H.; Barrett, D.C.; Biggs, A.; Eckersall, P.D. A peptidomic approach to biomarker discovery for bovine mastitis. J. Proteom. 2013, 85, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Peton, V.; Bouchard, D.S.; Almeida, S.; Rault, L.; Falentin, H.; Jardin, J.; Jan, G.; Hernandez, D.; François, P.; Schrenzel, J.; et al. Fine-tuned characterization of Staphylococcus aureus Newbould 305, a strain associated with mild and chronic mastitis in bovines. Vet. Res. 2014, 45, 106. [Google Scholar] [CrossRef]
- Siljamäki, P.; Varmanen, P.; Kankainen, M.; Pyörälä, S.; Karonen, T.; Iivanainen, A.; Auvinen, P.; Paulin, L.; Laine, P.K.; Taponen, S.; et al. Comparative proteome profiling of bovine and human Staphylococcus epidermidis strains for screening specifically expressed virulence and adaptation proteins. Proteomics 2014, 14, 1890–1894. [Google Scholar] [CrossRef] [PubMed]
- Poutanen, M.; Varhimo, E.; Kalkkinen, N.; Sukura, A.; Varmanen, P.; Savijoki, K. Two-dimensional difference gel electrophoresis analysis of Streptococcus uberis in response to mutagenesis-inducing ciprofloxacin challenge. J. Proteome Res. 2009, 8, 246–255. [Google Scholar] [CrossRef]
- Addis, M.F.; Pisanu, S.; Marogna, G.; Cubeddu, T.; Pagnozzi, D.; Cacciotto, C.; Campesi, F.; Schianchi, G.; Rocca, S.; Uzzau, S. Production and release of antimicrobial and immune defense proteins by mammary epithelial cells following Streptococcus uberis infection of sheep. Infect. Immun. 2013, 81, 3182–3197. [Google Scholar] [CrossRef]
- Pisanu, S.; Cubeddu, T.; Pagnozzi, D.; Rocca, S.; Cacciotto, C.; Alberti, A.; Marogna, G.; Uzzau, S.; Addis, M.F. Neutrophil extracellular traps in sheep mastitis. Vet. Res. 2015, 46, 59. [Google Scholar] [CrossRef] [PubMed]
- Lippolis, J.D.; Brunelle, B.W.; Reinhardt, T.A.; Sacco, R.E.; Nonnecke, B.J.; Dogan, B.; Simpson, K.; Schukken, Y.H. Proteomic analysis reveals protein expression differences in Escherichia coli strains associated with persistent versus transient mastitis. J. Proteom. 2014, 108, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.E.; Heller, E.D.; Sela, S.; Elad, D.; Edery, N.; Leitner, G. Genomic and Phenomic Study of Mammary pathogenic Escherichia coli. PLoS ONE 2015, 10, e0136387. [Google Scholar] [CrossRef] [PubMed]
- Katsafadou, A.I.; Tsangaris, G.T.; Anagnostopoulos, A.K.; Billinis, C.; Barbagianni, M.S.; Vasileiou, N.G.C.; Spanos, S.A.; Mavrogianni, V.S.; Fthenakis, G.C. Differential quantitative proteomics study of experimental Mannheimia haemolytica mastitis in sheep. J. Proteom. 2019, 205, 103393. [Google Scholar] [CrossRef] [PubMed]
- Katsafadou, A.I.; Tsangaris, G.T.; Vasileiou, N.G.C.; Ioannidi, K.S.; Anagnostopoulos, A.K.; Billinis, C.; Fragkou, I.A.; Papadopoulos, I.; Mavrogianni, V.S.; Michael, C.K.; et al. Detection of cathelicidin-1 in the milk as an early indicator of mastitis in ewes. Pathogens 2019, 8, 270. [Google Scholar] [CrossRef]
- Addis, M.F.; Pisanu, S.; Ghisaura, S.; Pagnozzi, D.; Marogna, G.; Tanca, A.; Biosa, G.; Cacciotto, C.; Alberti, A.; Pittau, M.; et al. Proteomics and pathway analyses of the milk fat globule in sheep naturally infected by Mycoplasma agalactiae provide indications of the in vivo response of the mammary epithelium to bacterial infection. Infect. Immun. 2011, 79, 3833–3845. [Google Scholar] [CrossRef]
- Katsafadou, A.I.; Vasileiou, N.G.C.; Fthenakis, G.C. Use of proteomics in the study of mastitis in ewes. Pathogens 2019, 8, 134. [Google Scholar] [CrossRef] [PubMed]
- Snelling, W.J.; Lin, Q.; Moore, J.E.; Millar, B.C.; Tosini, F.; Pozio, E.; Dooley, J.S.; Lowery, C.J. Proteomics analysis and protein expression during sporozoite excystation of Cryptosporidium parvum (Coccidia, Apicomplexa). Mol. Cell Proteom. 2007, 6, 346–355. [Google Scholar] [CrossRef]
- Sanderson, S.J.; Xia, D.; Prieto, H.; Yates, J.; Heiges, M.; Kissinger, J.C.; Bromley, E.; Lal, K.; Sinden, R.E.; Tomley, F.; et al. Determining the protein repertoire of Cryptosporidium parvum sporozoites. Proteomics 2008, 8, 1398–1414. [Google Scholar] [CrossRef]
- Sargison, N.D. Sheep Flock Health a Planned Approach; Blackwell Publishing: Oxford, UK, 2008; 476p. [Google Scholar]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef]
- Fritz, H.M.; Bowyer, P.W.; Bogyo, M.; Conrad, P.A.; Boothroyd, J.C. Proteomic analysis of fractionated Toxoplasma oocysts reveals clues to their environmental resistance. PLoS ONE 2012, 7, e29955. [Google Scholar] [CrossRef] [PubMed]
- Al-Bajalan, M.; Xia, D.; Armstrong, S.; Randle, N.; Wastling, J.M. Toxoplasma gondii and Neospora caninum induce different host cell responses at proteome-wide phosphorylation events; a step forward for uncovering the biological differences between these closely related parasites. Parasitol. Res. 2017, 116, 2707–2719. [Google Scholar] [CrossRef]
- Thompson, R.C.A. The zoonotic significance and molecular epidemiology of Giardia and giardiasis. Vet. Parasitol. 2004, 126, 15–35. [Google Scholar] [CrossRef]
- Thompson, R.C.; Monis, P. Giardia-from genome to proteome. Adv. Parasitol. 2012, 78, 57–95. [Google Scholar]
- Lauwaet, T.; Smith, A.J.; Reiner, D.S.; Romijn, E.P.; Wong, C.C.; Davids, B.J.; Shah, S.A.; Yates, J.R., 3rd; Gillin, F.D. Mining the Giardia genome and proteome for conserved and unique basal body proteins. Int. J. Parasitol. 2011, 41, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Jedelský, P.L.; Doležal, P.; Rada, P.; Pyrih, J.; Smíd, O.; Hrdý, I.; Sedinová, M.; Marcinčiková, M.; Voleman, L.; Perry, A.J.; et al. The minimal proteome in the reduced mitochondrion of the parasitic protist Giardia intestinalis. PLoS ONE 2011, 6, e17285. [Google Scholar] [CrossRef]
- Hagen, K.D.; Hirakawa, M.P.; House, S.A.; Schwartz, C.L.; Pham, J.K.; Cipriano, M.J.; De La Torre, M.J.; Sek, A.C.; Du, G.; Forsythe, B.M.; et al. Novel structural components of the ventral disc and lateral crest in Giardia intestinalis. PLoS Negl. Trop. Dis. 2011, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Coelho, C.H.; Costa, A.O.; Silva, A.C.; Pucci, M.M.; Serufo, A.V.; Busatti, H.G.; Durigan, M.; Perales, J.; Chapeaurouge, A.; da Silva, E.; et al. Genotyping and descriptive proteomics of a potential zoonotic canine strain of Giardia duodenalis, infective to mice. PLoS ONE 2016, 11, e0164946. [Google Scholar] [CrossRef] [PubMed]
- Capelli-Peixoto, J.; Mule, S.N.; Tano, F.T.; Palmisano, G.; Stolf, B.S. Proteomics and leishmaniasis: Potential clinical applications. Prot. Clin. 2019, 13, 1800136. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, Y.E.; Ouellette, M.; Papadopoulou, B. A proteomic approach to identify developmentally regulated proteins in Leishmania infantum. Proteomics 2002, 2, 1007–1017. [Google Scholar] [CrossRef]
- Zilberstein, D. Proteomic analysis of post-translational modifications using iTRAQ in Leishmania. Methods Mol. Biol. 2015, 1201, 261–268. [Google Scholar] [PubMed]
- Brotherton, M.C.; Bourassa, S.; Legare, D.; Poirier, G.G.; Droit, A.; Ouellette, M. Quantitative proteomic analysis of amphotericin B resistance in Leishmania infantum. Int. J. Parasitol. 2014, 4, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Drummelsmith, J.; Brochu, V.; Girard, I.; Messier, N.; Ouellette, M. Proteome mapping of the protozoan parasite Leishmania and application to the study of drug targets and resistance mechanisms. Mol. Cell. Proteom. 2003, 2, 146–155. [Google Scholar] [CrossRef]
- Morales, M.A.; Watanabe, R.; Dacher, M.; Chafey, P.; Osorio y Fortea, J.; Scott, D.A.; Beverley, S.M.; Ommen, G.; Clos, J.; Hem, S.; et al. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc. Natl. Acad. Sci. USA 2010, 107, 8381–8386. [Google Scholar] [CrossRef]
- Morales, M.A.; Watanabe, R.; Laurent, C.; Lenormand, P.; Rousselle, J.C.; Namane, A.; Spath, G.F. Phosphoproteomic analysis of Leishmania donovani pro- and amastigote stages. Proteomics 2008, 8, 350–363. [Google Scholar] [CrossRef]
- Garg, G.; Ali, V.; Singh, K.; Gupta, P.; Ganguly, A.; Sahasrabuddhe, A.A.; Das, P. Quantitative secretome analysis unravels new secreted proteins in Amphotericin B resistant Leishmania donovani. J. Proteom. 2019, 207, 103464. [Google Scholar] [CrossRef]
- Ejazi, S.A.; Bhattacharyya, A.; Choudhury, S.T.; Ghosh, S.; Sabur, A.; Pandey, K.; Das, V.; Das, P.; Rahaman, M.; Goswami, R.P.; et al. Immunoproteomic identification and characterization of Leishmania membrane proteins as non-invasive diagnostic candidates for clinical visceral leishmaniasis. Sci. Rep. 2018, 8, 12110. [Google Scholar] [CrossRef]
- Franco-Martínez, L.; Villar, M.; Tvarijonaviciute, A.; Escribano, D.; Bernal, L.J.; Cerón, J.J.; Thomas, M.; Mateos-Hernández, L.; Tecles, F.; de la Fuente, J.; et al. Serum proteome of dogs at subclinical and clinical onset of canine leishmaniosis. Transbound. Emerg. Dis. 2020, 67, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Agallou, M.; Athanasiou, E.; Samiotaki, M.; Panayotou, G.; Karagouni, E. Identification of Immunoreactive Leishmania infantum protein antigens to asymptomatic dog sera through combined immunoproteomics and bioinformatics analysis. PLoS ONE 2016, 11, e0149894. [Google Scholar] [CrossRef]
- Agallou, M.; Margaroni, M.; Kotsakis, S.D.; Karagouni, E. A Canine-directed chimeric multi-epitope vaccine induced protective immune responses in BALB/c mice infected with Leishmania infantum. Vaccines 2020, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Morante, T.; Shepherd, C.; Constantinoiu, C.; Loukas, A.; Sotillo, J. Revisiting the Ancylostoma caninum secretome provides new information on hookworm-host interactions. Proteomics 2017, 17, 1700186. [Google Scholar] [CrossRef]
- Richards, C.S.; Merritt, J.W. Studies on Angiostrongylus cantonensis in molluscan intermediate hosts. J. Parasitol. 1967, 53, 382–388. [Google Scholar] [CrossRef]
- Chen, K.Y.; Cheng, C.J.; Yen, C.M.; Tang, P.; Wang, L.C. Comparative studies on the proteomic expression patterns in the third- and fifth-stage larvae of Angiostrongylus cantonensis. Parasitol. Res. 2014, 113, 3591–3600. [Google Scholar] [CrossRef]
- Robinson, M.W.; Connolly, B. Proteomic analysis of the excretory-secretory proteins of the Trichinella spiralis L1 larva; a nematode parasite of skeletal muscle. Proteomics 2005, 5, 4525–4532. [Google Scholar] [CrossRef]
- Robinson, M.W.; Greig, R.; Beattie, K.A.; Lamont, D.J.; Connolly, B. Comparative analysis of the excretory-secretory proteome of the muscle larva of Trichinella pseudospiralis and Trichinella spiralis. Int. J. Parasitol. 2007, 37, 139–148. [Google Scholar] [CrossRef]
- Cui, J.; Liu, R.D.; Wang, L.; Zhang, X.; Jiang, P.; Liu, M.Y.; Wang, Z.Q. Proteomic analysis of surface proteins of Trichinella spiralis muscle larvae by two-dimensional gel electrophoresis and mass spectrometry. Parasit. Vectors 2013, 6, 355. [Google Scholar] [CrossRef]
- Liu, R.D.; Cui, J.; Liu, X.L.; Jiang, P.; Sun, G.G.; Zhang, X.; Long, S.R.; Wang, L.; Wang, Z.Q. Comparative proteomic analysis of surface proteins of Trichinella spiralis muscle larvae and intestinal infective larvae. Acta Trop. 2015, 150, 79–86. [Google Scholar] [CrossRef]
- Bien, J.; Cabaj, W.; Moskwa, B. Proteomic analysis of potential immunoreactive proteins from muscle larvae and adult worms of Trichinella spiralis in experimentally infected pigs. Folia Parasitol. 2015, 62, 022. [Google Scholar] [CrossRef] [PubMed]
- Gondek, M.; Herosimczyk, A.; Knysz, P.; Ożgo, M.; Lepczyński, A.; Szkucik, K. Comparative Proteomic Analysis of serum from pigs experimentally infected with Trichinella spiralis, Trichinella britovi, and Trichinella pseudospiralis. Pathogens 2020, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.; Zhang, W.; Li, J.; Loukas, A.; McManus, D.P.; Mulvenna, J. Proteomic characterisation of Echinococcus granulosus hydatid cyst fluid from sheep, cattle and humans. J. Proteom. 2011, 74, 1560–1572. [Google Scholar] [CrossRef]
- Ahn, C.S.; Kim, J.G.; Han, X.; Bae, Y.A.; Park, W.J.; Kang, I.; Wang, H.; Kong, Y. Biochemical Characterization of Echinococcus multilocularis antigen B3 reveals insight into adaptation and maintenance of parasitic homeostasis at the host-parasite interface. J. Proteome Res. 2017, 16, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, K.M.; Lorenzatto, K.R.; de Lima, J.C.; Dos Santos, G.B.; Förster, S.; Paludo, G.P.; Carvalho, P.C.; Brehm, K.; Ferreira, H.B. Comparative proteomics of hydatid fluids from two Echinococcus multilocularis isolates. J. Proteom. 2017, 162, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Zeghir-Bouteldja, R.; Polomé, A.; Bousbata, S.; Touil-Boukoffa, C. Comparative proteome profiling of hydatid fluid from Algerian patients reveals cyst location-related variation in Echinococcus granulosus. Acta Trop. 2017, 171, 199–206. [Google Scholar] [CrossRef]
- Ahn, C.S.; Han, X.; Bae, Y.A.; Ma, X.; Kim, J.T.; Cai, H.; Yang, H.J.; Kang, I.; Wang, H.; Kong, Y. Alteration of immunoproteome profile of Echinococcus granulosus hydatid fluid with progression of cystic echinococcosis. Parasit. Vectors 2015, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Vuitton, L.; Tuxun, T.; Li, J.; Vuitton, D.A.; Zhang, W.; McManus, D.P. Echinococcosis: Advances in the 21st century. Clin. Microbiol. Rev. 2019, 32, e00075-18. [Google Scholar] [CrossRef] [PubMed]
- Condell, O.; Sheridan, Á.; Power, K.A.; Bonilla-Santiago, R.; Sergeant, K.; Renaut, J.; Burgess, C.; Fanning, S.; Nally, J.E. Comparative proteomic analysis of Salmonella tolerance to the biocide active agent triclosan. J. Proteom. 2012, 75, 4505–4519. [Google Scholar] [CrossRef] [PubMed]
- Piras, C.; Soggiu, A.; Bonizzi, L.; Gaviraghi, A.; Deriu, F.; De Martino, L.; Iovane, G.; Amoresano, A.; Roncada, P. Comparative proteomics to evaluate multi drug resistance in Escherichia coli. Mol. Biosyst. 2012, 8, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Chernov, V.M.; Chernova, O.A.; Mouzykantov, A.A.; Lopukhov, L.L.; Aminov, R.I. Omics of antimicrobials and antimicrobial resistance. Expert Opin. Drug Discov. 2019, 14, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, W.; Zhang, R.; Xu, J.; Wang, R.; Wang, L.; Zhao, X.; Li, J. First acetyl-proteome profiling of Salmonella typhimurium revealed involvement of lysine acetylation in drug resistance. Vet. Microbiol. 2018, 226, 1–8. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Fu, Q.; Wang, Y.; Li, X.; Wu, C.; Shen, Z.; Zhang, Q.; Qin, P.; Shen, J.; et al. Integrated genomic and proteomic analyses of high-level chloramphenicol resistance in Campylobacter jejuni. Sci. Rep. 2017, 7, 16973. [Google Scholar] [CrossRef]
- Correia, S.; Nunes-Miranda, J.D.; Pinto, L.; Santos, H.M.; de Toro, M.; Sáenz, Y.; Torres, C.; Capelo, J.L.; Poeta, P.; Igrejas, G. Complete proteome of a quinolone-resistant Salmonella Typhimurium phage type DT104B clinical strain. Int. J. Mol. Sci. 2014, 15, 14191–14219. [Google Scholar] [CrossRef]
- Radford, D.; Strange, P.; Lepp, D.; Hernandez, M.; Rehman, M.A.; Diarra, M.S.; Balamurugan, S. Genomic and proteomic analyses of Salmonella enterica serovar Enteritidis identifying mechanisms of induced de novo tolerance to ceftiofur. Front. Microbiol. 2018, 9, 2123. [Google Scholar] [CrossRef]
- Wang, H.; McEntire, J.C.; Zhang, L.; Li, X.; Doyle, M. The transfer of antibiotic resistance from food to humans: Facts, implications and future directions. Rev. Sci. Tech. 2012, 31, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Loeffler, A.; Kadlec, K. Bacterial resistance to antimicrobial agents and its impact on veterinary and human medicine. Vet. Dermatol. 2017, 28, 82–95. [Google Scholar] [CrossRef]
- Vasileiou, N.; Chatzopoulos, D.C.; Sarrou, S.; Fragkou, I.A.; Katsafadou, A.I.; Mavrogianni, V.S.; Petinaki, E.; Fthenakis, G.C. Role of staphylococci in mastitis in sheep. J. Dairy Res. 2019, 86, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Taverna, F.; Negri, A.; Piccinini, R.; Zecconi, A.; Nonnis, S.; Ronchi, S.; Tedeschi, G. Characterization of cell wall associated proteins of a Staphylococcus aureus isolated from bovine mastitis case by a proteomic approach. Vet. Microbiol. 2007, 119, 240–247. [Google Scholar] [CrossRef]
- Liu, X.; Pai, P.J.; Zhang, W.; Hu, Y.; Dong, X.; Qian, P.Y.; Chen, D.; Lam, H. Proteomic response of methicillin-resistant S. aureus to a synergistic antibacterial drug combination: A novel erythromycin derivative and oxacillin. Sci. Rep. 2016, 6, 19841. [Google Scholar] [CrossRef] [PubMed]
- Igrejas, G.; Correia, S.; Silva, V.; Hébraud, M.; Caniça, M.; Torres, C.; Gomes, C.; Nogueira, F.; Poeta, P. Planning a one health case study to evaluate methicillin resistant Staphylococcus aureus and its economic burden in Portugal. Front. Microbiol. 2018, 9, 2964. [Google Scholar] [CrossRef]
- Monteiro, R.; Vitorino, R.; Domingues, P.; Radhouani, H.; Carvalho, C.; Poeta, P.; Torres, C.; Igrejas, G. Proteome of a methicillin-resistant Staphylococcus aureus clinical strain of sequence type ST398. J. Proteom. 2012, 75, 2892–2915. [Google Scholar] [CrossRef]
- Abril, A.G.; Carrera, M.; Böhme, K.; Barros-Velázquez, J.; Rama, J.R.; Calo-Mata, P.; Sánchez-Pérez, A.; Villa, T.G. Proteomic characterization of antibiotic resistance, and production of antimicrobial and virulence factors in Streptococcus species associated with bovine mastitis. Could enzybiotics represent novel therapeutic agents against these pathogens? Antibiotics 2020, 9, 302. [Google Scholar] [CrossRef]
- Ramos, S.; Chafsey, I.; Hebraud, M.; Sousa, M.; Poeta, P.; Igrejas, G. Ciprofloxacin sress proteome of the extended-spectrum beta-lactamase producing Escherichia coli from slaughtered pigs. Curr. Proteom. 2016, 13, 285. [Google Scholar] [CrossRef]
- Piras, C.; Soggiu, A.; Greco, V.; Martino, P.A.; Del Chierico, F.; Putignani, L.; Urbani, A.; Nally, J.E.; Bonizzi, L.; Roncada, P. Mechanisms of antibiotic resistance to enrofloxacin in uropathogenic Escherichia coli in dog. J. Proteom. 2015, 127, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, D.F.; Lin, X.M.; Peng, X.X. Outer membrane proteomics of kanamycin-resistant Escherichia coli identified MipA as a novel antibiotic resistance-related protein. FEMS Microbiol. Lett. 2015, 362, fnv074. [Google Scholar]
- Nanduri, B.; Lawrence, M.L.; Vanguri, S.; Pechan, T.; Burgess, S.C. Proteomic analysis using an unfinished bacterial genome: The effects of subminimum inhibitory concentrations of antibiotics on Mannheimia haemolytica virulence factor expression. Proteomics 2005, 5, 4852–4863. [Google Scholar] [CrossRef] [PubMed]
- Mozzarelli, A.; Paredi, G.; Pioselli, B.; Raboni, S. From meat to food: The proteomics assessment. In Farm Animal Proteomics 2012, Proceedings of the 3rd Managing Committee Meeting and 2nd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Algarve, Portugal, 27 March 2012; Rodrigues, P., Eckersall, D., de Almeida, A., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2012; pp. 30–34. [Google Scholar]
- Roncada, P.; Soggiu, A.; Piras, C.; Bonizzi, L. Microbial proteomics in food safety and animal welfare. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 38–39. [Google Scholar]
- Ferranti, P.; Roncada, P.; Scaloni, A. Foodomics—Novel insights in food and nutrition domains. J. Proteom. 2016, 147, 1–2. [Google Scholar] [CrossRef]
- Sospedra, I.; Soriano, J.M.; Mañes, J. Enterotoxinomics: The omic sciences in the study of staphylococcal toxins analyzed in food matrices. Food Res. Int. 2013, 54, 1052–1060. [Google Scholar] [CrossRef]
- Marzano, V.; Tilocca, B.; Fiocchi, A.G.; Vernocchi, P.; Levi Mortera, S.; Urbani, A.; Roncada, P.; Putignani, L. Perusal of food allergens analysis by mass spectrometry-based proteomics. J. Proteom. 2020, 215, 103636. [Google Scholar] [CrossRef]
- Piras, C.; Roncada, P.; Rodrigues, P.M.; Bonizzi, L.; Soggiu, A. Proteomics in food: Quality, safety, microbes, and allergens. Proteomics 2016, 16, 799–815. [Google Scholar] [CrossRef]
- Piñeiro, C.; Barros-Velázquez, J.; Vázquez, J.; Figueras, A.; Gallardo, J.M. Proteomics as a tool for the investigation of seafood and other marine products. J. Proteome Res. 2003, 2, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, S.; Mullen, W.; Mischak, H.; Bradshaw, C.; Cristobal, S. Shotgun proteomics in blue mussels exposed to benthic trawler-induced sediment resuspension from a polluted fjord. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 258–261. [Google Scholar]
- Arroyo, L.; Marco-Ramell, A.; Soler, M.; Pena, R.; Velarde, A.; Sabria, J.; Unzeta, M.; Bassols, A. Neurotransmitter levels and proteomic approach in pig brain: Pre-slaughter handling stress and cognitive biases. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 192–195. [Google Scholar]
- Clerens, S.; Plowman, J.E.; Haines, S.; Dyer, J.M. A mass spectrometric scoring system for oxidative damage in dairy foods. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 255–257. [Google Scholar]
- Soggiu, A.; Bendixen, E.; Brasca, M.; Morandi, S.; Piras, C.; Bonizzi, L.; Roncada, P. Milk and cheese microbiome for safety and quality of dairy products. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 262–265. [Google Scholar]
- Marrocco, C.; D’Alessandro, A.; Rinalducci, S.; Mirasole, C.; Zolla, L. Untargeted metabolomic analyses open new scenarios in post mortem pig muscles: Casertan and Large White. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 270–273. [Google Scholar]
- Paredi, G.; Benoni, R.; Pighini, G.; Ronda, L.; Dowle, A.; Ashford, D.; Thomas, J.; Saccani, G.; Virgili, R.; Mozzarelli, A. Proteomics of Parma dry-cured ham: Analysis of salting exudates. J. Agric. Food Chem. 2017, 65, 6307–6316. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, H.; Cao, C.; Zhao, W.; Kwok, L.Y.; Zhang, H.; Zhang, W. Characterization of the adaptive amoxicillin resistance of Lactobacillus casei Zhang by proteomic analysis. Front. Microbiol. 2018, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Rinalducci, S.; Marrocco, C.; Zolla, V.; Napolitano, F.; Zolla, L. Love me tender: An Omics window on the bovine meat tenderness network. J. Proteom. 2012, 75, 4360–4380. [Google Scholar] [CrossRef]
- Bonnet, M.; Kaspric, N.; Picard, B. Quest for biomarkers of the lean-to-fat ratio by proteomics in beef production. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 43–44. [Google Scholar]
- Di Luca, A.; Elia, G.; Hamill, R.; Mullen, A.M. 2D DIGE proteomic analysis of early post mortem muscle exudate highlights the importance of the stress response for improved water-holding capacity of fresh pork meat. Proteomics 2013, 13, 1528–1544. [Google Scholar] [CrossRef]
- Hamelin, M.; Sayd, T.; Chambon, C.; Bouix, J.; Bibé, B.; Milenkovic, D.; Leveziel, H.; Georges, M.; Clop, A.; Marinova, P.; et al. Proteomic analysis of ovine muscle hypertrophy. J. Anim. Sci. 2006, 84, 3266–3276. [Google Scholar] [CrossRef][Green Version]
- Paredi, G.; Raboni, S.; Bendixen, E.; de Almeida, A.M.; Mozzarelli, A. “Muscle to meat” molecular events and technological transformations: The proteomics insight. J. Proteom. 2012, 75, 4275–4289. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Restelli, L.; Araujo, S.S.; Dilda, F.; Sales-Baptista, E.; Ceciliani, F.; Almeida, A.M. Bitter taste in water-buffalo (Bubalus bubalis): From T2R gene identification to expression studies. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 199–203. [Google Scholar]
- D’Alessandro, A.; Righetti, P.G.; Fasoli, E.; Zolla, L. The egg white and yolk interactomes as gleaned from extensive proteomic data. J. Proteom. 2010, 73, 1028–1042. [Google Scholar] [CrossRef]
- Omana, D.A.; Liang, Y.; Kav, N.N.; Wu, J. Proteomic analysis of egg white proteins during storage. Proteomics 2011, 11, 144–153. [Google Scholar] [CrossRef]
- Anagnostopoulos, A.K.; Tsangaris, G.T. Feta cheese proteins: Manifesting the identity of Greece’s national treasure. Data Brief 2019, 19, 2037–2040. [Google Scholar] [CrossRef]
- Scarselli, R.; Donadio, E.; Giuffrida, M.G.; Fortunato, D.; Conti, A.; Balestreri, E.; Felicioli, R.; Pinzauti, M.; Sabatini, A.G.; Felicioli, A. Towards royal jelly proteome. Proteomics 2005, 5, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.B.; Attard, E.; Camilleri, C. Molecular characterization of Maltese honey: Diastase and proline levels changes in Maltese honey seasons. In Farm Animal Proteomics 2013, Proceedings of the 4th Management Committee Meeting and 3rd Meeting of Working Groups 1, 2 & 3 of COST Action FA1002, Kosice, Slovakia, 25–26 April 2013; de Almeida, A., Eckersall, D., Bencurova, E., Dolinska, S., Mlynarcik, P., Vincova, M., Bhide, M.R., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2013; pp. 266–269. [Google Scholar]
- Cain, J.A.; Dale, A.L.; Niewold, P.; Klare, W.P.; Man, L.; White, M.Y.; Scott, N.E.; Cordwell, S.J. Proteomics reveals multiple phenotypes associated with N-linked glycosylation in Campylobacter jejuni. Mol. Cell. Proteom. 2019, 18, 715–734. [Google Scholar] [CrossRef]
- Sabença, C.; Sousa, T.; Oliveira, S.; Viala, D.; Théron, L.; Chambon, C.; Hébraud, M.; Beyrouthy, R.; Bonnet, R.; Caniça, M.; et al. Next-generation sequencing and MALDI mass spectrometry in the study of multiresistant processed meat vancomycin-resistant enterococci (VRE). Biology 2020, 9, 89. [Google Scholar] [CrossRef]
- Witt, N.; Andreotti, S.; Busch, A.; Neubert, K.; Reinert, K.; Tomaso, H.; Meierhofer, D. Rapid and culture free identification of Francisella in hare carcasses by high-resolution tandem mass spectrometry proteotyping. Front. Microbiol. 2020, 11, 636. [Google Scholar] [CrossRef]
- Jadhav, S.R.; Shah, R.M.; Karpe, A.V.; Morrison, P.D.; Kouremenos, K.; Beale, D.J.; Palombo, E.A. Detection of foodborne pathogens using proteomics and metabolomics-based approaches. Front. Microbiol. 2018, 9, 3132. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yang, L.; Yu, P.; Wang, Y.; Peng, X.; Chen, L. Comparative proteomics and secretomics revealed virulence and antibiotic resistance-associated factors in Vibrio parahaemolyticus recovered from commonly consumed aquatic products. Front. Microbiol. 2020, 11, 1453. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.D.; Letellier, A.; Beaudry, F. Analysis of Staphylococcus enterotoxin B using differential isotopic tags and liquid chromatography quadrupole ion trap mass spectrometry. Biomed. Chromatogr. 2012, 26, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, M.; Tsilia, V.; Rajkovic, A.; De Cremer, K.; Van Loco, J. Application of LC-MS/MS MRM to determine staphylococcal enterotoxins (SEB and SEA) in milk. Toxins 2016, 8, 118. [Google Scholar] [CrossRef]
- Saleh, S.; Staes, A.; Deborggraeve, S.; Gevaert, K. Targeted proteomics for studying pathogenic bacteria. Proteomics 2019, 19, e1800435. [Google Scholar] [CrossRef]
- Dupré, M.; Gilquin, B.; Fenaille, F.; Feraudet-Tarisse, C.; Dano, J.; Ferro, M.; Simon, S.; Junot, C.; Brun, V.; Becher, F. Multiplex quantification of protein toxins in human biofluids and food matrices using immunoextraction and high-resolution targeted mass spectrometry. Anal. Chem. 2015, 87, 8473–8480. [Google Scholar] [CrossRef] [PubMed]
- Gilquin, B.; Jaquinod, M.; Louwagie, M.; Kieffer-Jaquinod, S.; Kraut, A.; Ferro, M.; Becher, F.; Brun, V. A proteomics assay to detect eight CBRN-relevant toxins in food. Proteomics 2017, 17, 1600357. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.; Kim, D.; Jung, K.; Kang, S.; Lee, Y.H. Fungal secretome database: Integrated platform for annotation of fungal secretomes. BMC Genom. 2010, 11, 105. [Google Scholar] [CrossRef]
- Bhadauria, V.; Banniza, S.; Wang, L.X.; Wei, Y.D.; Peng, Y.L. Proteomics studies of phytopathogenic fungi, oomycetes and their interactions with hosts. Eur. J. Plant. Pathol. 2009, 126, 81–95. [Google Scholar] [CrossRef]
- Nzoughet, K.J.; Hamilton, J.T.; Floyd, S.D.; Douglas, A.; Nelson, J.; Devine, L.; Elliott, C.T. Azaspiracid: First evidence of protein binding in shellfish. Toxicon 2008, 51, 1255–1263. [Google Scholar] [CrossRef]
- Nzoughet, J.K.; Hamilton, J.T.; Botting, C.H.; Douglas, A.; Devine, L.; Nelson, J.; Elliott, C.T. Proteomics identification of azaspiracid toxin biomarkers in blue mussels, Mytilus edulis. Mol. Cell. Proteom. 2009, 8, 1811–1822. [Google Scholar] [CrossRef]
- Bu, G.; Luo, Y.; Chen, F.; Liu, K.; Zhu, T. Milk processing as a tool to reduce cow’s milk allergenicity: A mini-review. Dairy Sci. Technol. 2013, 93, 211–223. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, E.; Mameli, C.; Piras, C.; Cococcioni, L.; Urbani, A.; Zuccotti, G.V.; Roncada, P. Precision medicine in cow’s milk allergy: Proteomics perspectives from allergens to patients. J. Proteom. 2018, 188, 173–180. [Google Scholar] [CrossRef]
- Carrera, M.; Cañas, B.; Gallardo, J.M. Advanced proteomics and systems biology applied to study food allergy. Curr. Opin. Food Sci. 2018, 22, 9–16. [Google Scholar] [CrossRef]
- Carrera, M.; Cañas, B.; Gallardo, J.M. Rapid direct detection of the major fish allergen, parvalbumin, by selected MS/MS ion monitoring mass spectrometry. J. Proteom. 2012, 75, 3211–3220. [Google Scholar] [CrossRef]
- Abdel Rahman, A.M.; Kamath, S.; Lopata, A.L.; Helleur, R.J. Analysis of the allergenic proteins in black tiger prawn (Penaeus monodon) and characterization of the major allergen tropomyosin using mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Nardiello, D.; Natale, A.; Palermo, C.; Quinto, M.; Centonze, D. Milk authenticity by ion-trap proteomics following multi-enzyme digestion. Food Chem. 2018, 244, 317–323. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsarou, E.I.; Billinis, C.; Galamatis, D.; Fthenakis, G.C.; Tsangaris, G.T.; Katsafadou, A.I. Applied Proteomics in ‘One Health’. Proteomes 2021, 9, 31. https://doi.org/10.3390/proteomes9030031
Katsarou EI, Billinis C, Galamatis D, Fthenakis GC, Tsangaris GT, Katsafadou AI. Applied Proteomics in ‘One Health’. Proteomes. 2021; 9(3):31. https://doi.org/10.3390/proteomes9030031
Chicago/Turabian StyleKatsarou, Eleni I., Charalambos Billinis, Dimitrios Galamatis, George C. Fthenakis, George Th. Tsangaris, and Angeliki I. Katsafadou. 2021. "Applied Proteomics in ‘One Health’" Proteomes 9, no. 3: 31. https://doi.org/10.3390/proteomes9030031
APA StyleKatsarou, E. I., Billinis, C., Galamatis, D., Fthenakis, G. C., Tsangaris, G. T., & Katsafadou, A. I. (2021). Applied Proteomics in ‘One Health’. Proteomes, 9(3), 31. https://doi.org/10.3390/proteomes9030031