Misincorporation Proteomics Technologies: A Review

 ,
,  , , and
, , and 
Abstract
1. Introduction
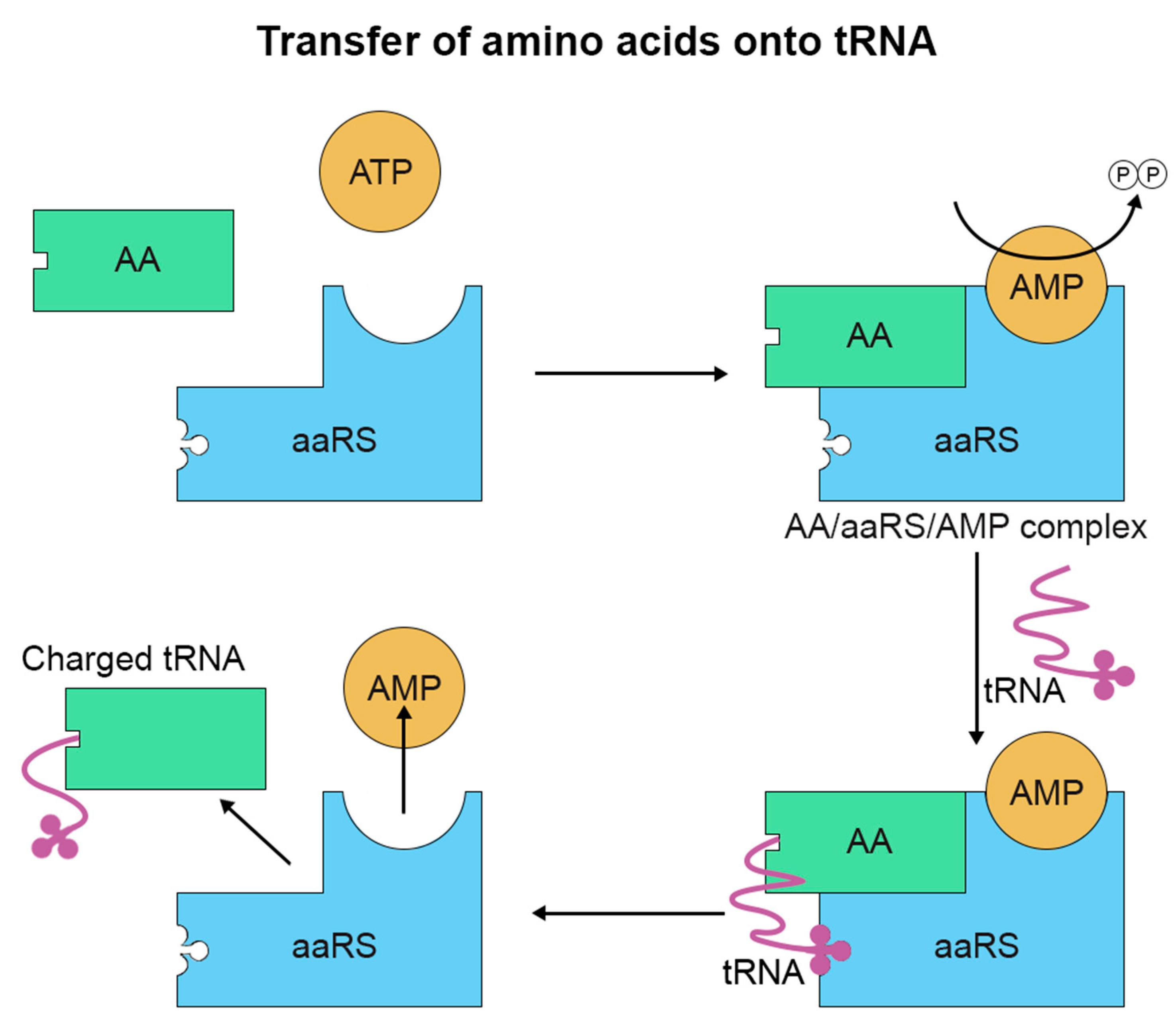
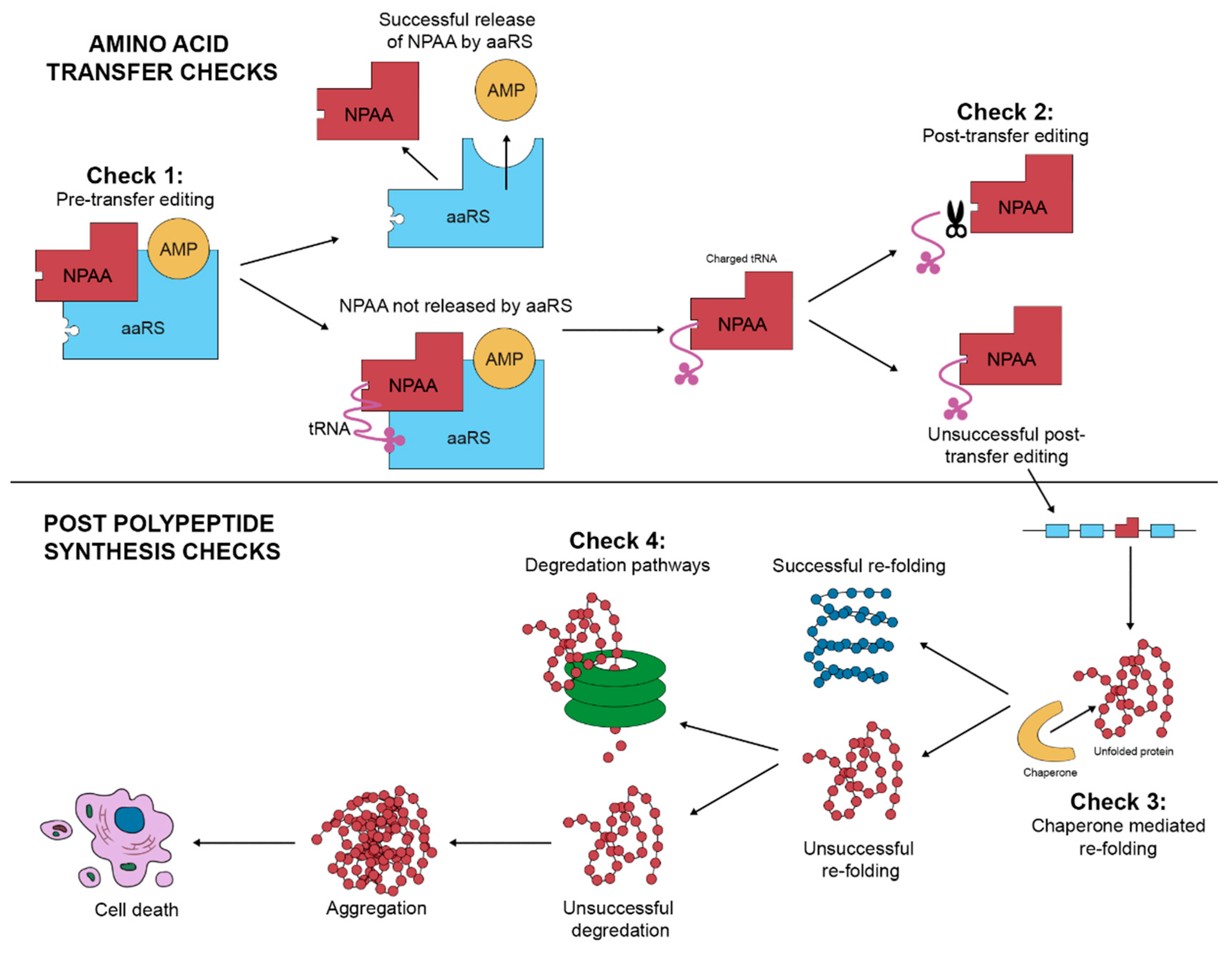
2. Amino Acid Misincorporation
3. The Identification of NPAAs Misincorporation
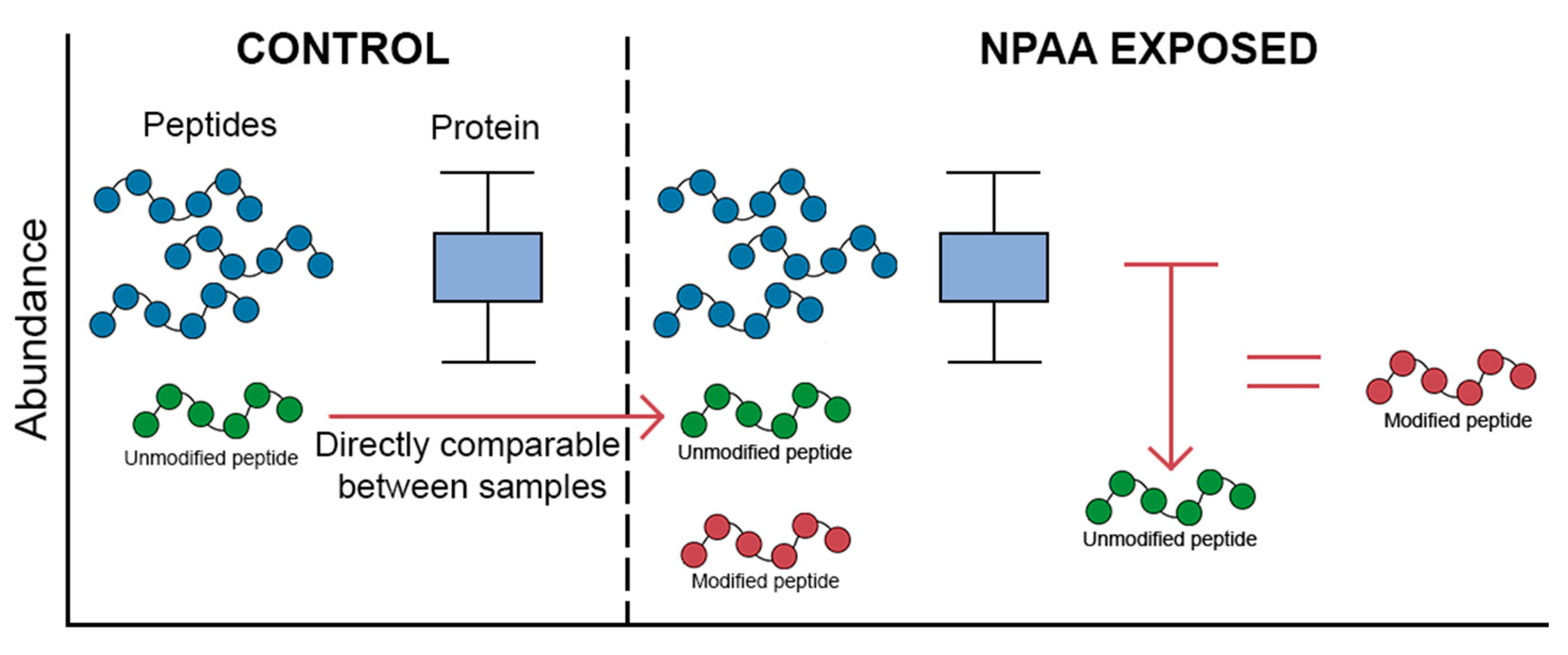
4. Key Considerations in Mistranslation Proteomics
5. Sample Processing and Enrichment
6. Mass Spectrometric Technologies
7. Mass Spectrometer Base Requirements and Desirable Features
8. Data Dependent Analysis
9. Data Independent Acquisition
10. Immonium Ion and Precursor Ion Scanning
11. Ion Mobility Mass Spectrometry
12. When Is an Incorporation Real?
13. Data Analysis Techniques
14. Conclusions, a Future Direction and a Best Practice for MiP
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down, P. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Shliaha, P.V.; Gorshkov, V.; Kovalchuk, S.I.; Schwammle, V.; Baird, M.A.; Shvartsburg, A.A.; Jensen, O.N. Middle-Down Proteomic Analyses with Ion Mobility Separations of Endogenous Isomeric Proteoforms. Anal. Chem. 2020, 92, 2364–2368. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, C.; Boll, I.; Finsen, B.; Modzel, M.; Larsen, M.R. Characterizing disease-associated changes in post-translational modifications by mass spectrometry. Expert Rev. Proteom. 2018, 15, 245–258. [Google Scholar] [CrossRef]
- Berry, I.J.; Steele, J.R.; Padula, M.P.; Djordjevic, S.P. The application of terminomics for the identification of protein start sites and proteoforms in bacteria. Proteomics 2016, 16, 257–272. [Google Scholar] [CrossRef]
- Mohler, K.; Ibba, M. Translational fidelity and mistranslation in the cellular response to stress. Nat. Microbiol. 2017, 2, 17117. [Google Scholar] [CrossRef]
- Garofalo, R.; Wohlgemuth, I.; Pearson, M.; Lenz, C.; Urlaub, H.; Rodnina, M.V. Broad range of missense error frequencies in cellular proteins. Nucleic. Acids Res. 2019, 47, 2932–2945. [Google Scholar] [CrossRef]
- Drummond, D.A.; Wilke, C.O. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef]
- Jarocki, V.M.; Steele, J.R.; Widjaja, M.; Tacchi, J.L.; Padula, M.P.; Djordjevic, S.P. Formylated N-terminal methionine is absent from the Mycoplasma hyopneumoniae proteome: Implications for translation initiation. Int. J. Med. Microbiol. 2019, 309, 288–298. [Google Scholar] [CrossRef]
- Mordret, E.; Dahan, O.; Asraf, O.; Rak, R.; Yehonadav, A.; Barnabas, G.D.; Cox, J.; Geiger, T.; Lindner, A.B.; Pilpel, Y. Systematic Detection of Amino Acid Substitutions in Proteomes Reveals Mechanistic Basis of Ribosome Errors and Selection for Translation Fidelity. Mol. Cell 2019, 75, 427–441e425. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.A.; Wilke, C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.J.; Garcia-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.A. Nonprotein amino acids of plants: Significance in medicine, nutrition, and agriculture. J. Agric. Food Chem. 2003, 51, 2854–2865. [Google Scholar] [CrossRef]
- Chan, S.W.; Dunlop, R.A.; Rowe, A.; Double, K.L.; Rodgers, K.J. l-DOPA is incorporated into brain proteins of patients treated for Parkinson’s disease, inducing toxicity in human neuroblastoma cells in vitro. Exp. Neurol. 2012, 238, 29–37. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Spencer, P.S.; Nunn, P.B.; Hugon, J.; Ludolph, A.C.; Ross, S.M.; Roy, D.N.; Robertson, R.C. Guam Amyotrophic-Lateral-Sclerosis Parkinsonism Dementia Linked to a Plant Excitant Neurotoxin. Science 1987, 237, 517–522. [Google Scholar] [CrossRef]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar] [CrossRef]
- Rubenstein, E. Misincorporation of the Proline Analog Azetidine-2-Carboxylic Acid in the Pathogenesis of Multiple Sclerosis: A Hypothesis. J. Neuropathol. Exp. Neurol. 2008, 67, 1035–1040. [Google Scholar] [CrossRef]
- Bessonov, K.; Bamm, V.V.; Harauz, G. Misincorporation of the proline homologue Aze (azetidine-2-carboxylic acid) into recombinant myelin basic protein. Phytochemistry 2010, 71, 502–507. [Google Scholar] [CrossRef]
- Ravindranath, V. Neurolathyrism: Mitochondrial dysfunction in excitotoxicity mediated by L-beta-oxalyl aminoalanine. Neurochem. Int. 2002, 40, 505–509. [Google Scholar] [CrossRef]
- Yan, Z.Y.; Spencer, P.S.; Li, Z.X.; Liang, Y.M.; Wang, Y.F.; Wang, C.Y.; Li, F.M. Lathyrus sativus (grass pea) and its neurotoxin ODAP. Phytochemistry 2006, 67, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, V.; Kalita, P.; Shukla, H.; Kumar, A.; Tripathi, T. Aminoacyl-tRNA synthetases: Structure, function, and drug discovery. Int. J. Biol. Macromol. 2018, 111, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Bullwinkle, T.; Lazazzera, B.; Ibba, M. Quality Control and Infiltration of Translation by Amino Acids Outside of the Genetic Code. Annu. Rev. Genet. 2014, 48, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Mohler, K.; Aerni, H.R.; Gassaway, B.; Ling, J.; Ibba, M.; Rinehart, J. MS-READ: Quantitative measurement of amino acid incorporation. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3081–3088. [Google Scholar] [CrossRef]
- Ling, J.; Reynolds, N.; Ibba, M. Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 2009, 63, 61–78. [Google Scholar] [CrossRef]
- Giannopoulos, S.; Samardzic, K.; Raymond, B.B.A.; Djordjevic, S.P.; Rodgers, K.J. L-DOPA causes mitochondrial dysfunction in vitro: A novel mechanism of L-DOPA toxicity uncovered. Int. J. Biochem. Cell Biol. 2019, 117, 105624. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Cox, P.A.; Banack, S.A.; Rodgers, K.J. The non-protein amino acid BMAA is misincorporated into human proteins in place of L-serine causing protein misfolding and aggregation. PLoS ONE 2013, 8, e75376. [Google Scholar] [CrossRef]
- Rubenstein, E.; Zhou, H.L.; Krasinska, K.M.; Chien, A.; Becker, C.H. Azetidine-2-carboxylic acid in garden beets (Beta vulgaris). Phytochemistry 2006, 67, 898–903. [Google Scholar] [CrossRef]
- Cox, P.A.; Davis, D.A.; Mash, D.C.; Metcalf, J.S.; Banack, S.A. Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc. Biol. Sci. 2016, 283. [Google Scholar] [CrossRef]
- Rodgers, K.J. Non-protein amino acids and neurodegeneration: The enemy within. Exp. Neurol. 2014, 253, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Richmond, M.H. The effect of amino acid analogues on growth and protein synthesis in microorganisms. Bacteriol. Rev. 1962, 26, 398–420. [Google Scholar] [CrossRef] [PubMed]
- Fowden, L.; Lewis, D.; Tristram, H. Toxic amino acids: Their action as antimetabolites. Adv. Enzymol. Relat. Areas Mol. Biol. 1967, 29, 89–163. [Google Scholar] [CrossRef] [PubMed]
- Fowden, L.; Richmond, M.H. Replacement of proline by azetidine-2-carboxylic acid during biosynthesis of protein. Biochim. Biophys. Acta 1963, 71, 459–461. [Google Scholar] [CrossRef]
- Fowden, L. Nitrogenous compounds and nitrogen metabolism in the Liliaceae. 6. Changes in nitrogenous composition during the growth of Convallaria and Polygonatum. Biochem. J. 1959, 71, 643–648. [Google Scholar] [CrossRef]
- Richmond, M.H. Incorporation of canavanine by Staphylococcus aureus 524 SC. Biochem. J. 1959, 73, 261–264. [Google Scholar] [CrossRef]
- Walker, J.B. Canavanine and homoarginine as antimetabolites of arginine and lysine in yeast and algae. J. Biol. Chem. 1955, 212, 207–215. [Google Scholar] [CrossRef]
- Schwartz, J.H.; Maas, W.K. Analysis of the inhibition of growth produced by canavanine in Escherichia coli. J. Bacteriol. 1960, 79, 794–799. [Google Scholar] [CrossRef]
- Aronson, J.N.; Wermus, G.R. Effects of M-Tyrosine on Growth and Sporulation of Bacillus Species. J. Bacteriol. 1965, 90, 38–46. [Google Scholar] [CrossRef]
- Netzer, N.; Goodenbour, J.M.; David, A.; Dittmar, K.A.; Jones, R.B.; Schneider, J.R.; Boone, D.; Eves, E.M.; Rosner, M.R.; Gibbs, J.S.; et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462, 522–526. [Google Scholar] [CrossRef]
- Main, B.J.; Bowling, L.C.; Padula, M.P.; Bishop, D.P.; Mitrovic, S.M.; Guillemin, G.J.; Rodgers, K.J. Detection of the suspected neurotoxin beta-methylamino-l-alanine (BMAA) in cyanobacterial blooms from multiple water bodies in Eastern Australia. Harmful Algae 2018, 74, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Han, N.C.; Bullwinkle, T.J.; Loeb, K.F.; Faull, K.F.; Mohler, K.; Rinehart, J.; Ibba, M. The mechanism of beta-N-methylamino-l-alanine inhibition of tRNA aminoacylation and its impact on misincorporation. J. Biol. Chem. 2020, 295, 1402–1410. [Google Scholar] [CrossRef]
- Edelmann, P.; Gallant, J. Mistranslation in E. coli. Cell 1977, 10, 131–137. [Google Scholar] [CrossRef]
- Bouadloun, F.; Donner, D.; Kurland, C.G. Codon-specific missense errors in vivo. EMBO J. 1983, 2, 1351–1356. [Google Scholar] [CrossRef]
- Khazaie, K.; Buchanan, J.H.; Rosenberger, R.F. The accuracy of Qβ RNA translation. Eur. J. Biochem. 1984, 144, 491–495. [Google Scholar] [CrossRef]
- Loftfield, R.B.; Vanderjagt, D. The frequency of errors in protein biosynthesis. Biochem. J. 1972, 128, 1353–1356. [Google Scholar] [CrossRef]
- Parker, J.; Friesen, J.D. “Two out of three” codon reading leading to mistranslation in vivo. MGG Mol. Gen. Genet. 1980, 177, 439–445. [Google Scholar] [CrossRef]
- Toth, M.J.; Murgola, E.J.; Schimmel, P. Evidence for a unique first position codon-anticodon mismatch in vivo. J. Mol. Biol. 1988, 201, 451–454. [Google Scholar] [CrossRef]
- Stansfield, I.; Jones, K.M.; Herbert, P.; Lewendon, A.; Shaw, W.V.; Tuite, M.F. Missense translation errors in Saccharomyces cerevisiae11Edited by K. Nagai. J. Mol. Biol. 1998, 282, 13–24. [Google Scholar] [CrossRef]
- Curran, J.F.; Yarus, M. Base substitutions in the tRNA anticodon arm do not degrade the accuracy of reading frame maintenance. Proc. Natl. Acad. Sci. USA 1986, 83, 6538. [Google Scholar] [CrossRef]
- Meyerovich, M.; Mamou, G.; Ben-Yehuda, S. Visualizing high error levels during gene expression in living bacterial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11543–11548. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.B.; Farabaugh, P.J. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 2007, 13, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Javid, B.; Sorrentino, F.; Toosky, M.; Zheng, W.; Pinkham, J.T.; Jain, N.; Pan, M.; Deighan, P.; Rubin, E.J. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Ribas de Pouplana, L.; Santos, M.A.S.; Zhu, J.-H.; Farabaugh, P.J.; Javid, B. Protein mistranslation: Friend or foe? Trends Biochem. Sci. 2014, 39, 355–362. [Google Scholar] [CrossRef]
- Ruan, B.; Palioura, S.; Sabina, J.; Marvin-Guy, L.; Kochhar, S.; LaRossa, R.A.; Söll, D. Quality control despite mistranslation caused by an ambiguous genetic code. Proc. Natl. Acad. Sci. USA 2008, 105, 16502–16507. [Google Scholar] [CrossRef]
- Yu, X.C.; Borisov, O.V.; Alvarez, M.; Michels, D.A.; Wang, Y.J.; Ling, V. Identification of Codon-Specific Serine to Asparagine Mistranslation in Recombinant Monoclonal Antibodies by High-Resolution Mass Spectrometry. Anal. Chem. 2009, 81, 9282–9290. [Google Scholar] [CrossRef]
- Ozawa, K.; Headlam, M.J.; Mouradov, D.; Watt, S.J.; Beck, J.L.; Rodgers, K.J.; Dean, R.T.; Huber, T.; Otting, G.; Dixon, N.E. Translational incorporation of L-3,4-dihydroxyphenylalanine into proteins. FEBS J. 2005, 272, 3162–3171. [Google Scholar] [CrossRef]
- Creasy, D.M.; Cottrell, J.S. Unimod: Protein modifications for mass spectrometry. Proteomics 2004, 4, 1534–1536. [Google Scholar] [CrossRef]
- Samardzic, K.; Rodgers, K.J. Cytotoxicity and mitochondrial dysfunction caused by the dietary supplement l-norvaline. Toxicol. In Vitro 2019, 56, 163–171. [Google Scholar] [CrossRef]
- Samardzic, K.; Rodgers, K.J. Cell death and mitochondrial dysfunction induced by the dietary non-proteinogenic amino acid L-azetidine-2-carboxylic acid (Aze). Amino Acids 2019, 51, 1221–1232. [Google Scholar] [CrossRef]
- Svinkina, T.; Gu, H.; Silva, J.C.; Mertins, P.; Qiao, J.; Fereshetian, S.; Jaffe, J.D.; Kuhn, E.; Udeshi, N.D.; Carr, S.A. Deep, Quantitative Coverage of the Lysine Acetylome Using Novel Anti-acetyl-lysine Antibodies and an Optimized Proteomic Workflow. Mol. Cell. Proteom. MCP 2015, 14, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.R.; Trelle, M.B.; Thingholm, T.E.; Jensen, O.N. Analysis of posttranslational modifications of proteins by tandem mass spectrometry. Biotechniques 2006, 40, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Koide, S. Next-generation antibodies for post-translational modifications. Curr. Opin. Struct. Biol. 2018, 51, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef] [PubMed]
- Gozal, Y.M.; Duong, D.M.; Gearing, M.; Cheng, D.; Hanfelt, J.J.; Funderburk, C.; Peng, J.; Lah, J.J.; Levey, A.I. Proteomics analysis reveals novel components in the detergent-insoluble subproteome in Alzheimer’s disease. J. Proteome Res. 2009, 8, 5069–5079. [Google Scholar] [CrossRef] [PubMed]
- van Onselen, R.; Cook, N.A.; Phelan, R.R.; Downing, T.G. Bacteria do not incorporate β-N-methylamino-l-alanine into their proteins. Toxicon 2015, 102, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Quantitative Analysis of in Vivo Methionine Oxidation of the Human Proteome. J. Proteome Res. 2020, 19, 624–633. [Google Scholar] [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A.; et al. Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat. Methods 2019, 16, 509–518. [Google Scholar] [CrossRef]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Aggarwal, K.; Choe, L.H.; Lee, K.H. Shotgun proteomics using the iTRAQ isobaric tags. Brief. Funct. Genom. 2006, 5, 112–120. [Google Scholar] [CrossRef]
- Beri, J.; Nash, T.; Martin, R.M.; Bereman, M.S. Exposure to BMAA mirrors molecular processes linked to neurodegenerative disease. Proteomics 2017, 17, 10. [Google Scholar] [CrossRef]
- Hecht, E.S.; Scigelova, M.; Eliuk, S.; Makarov, A. Fundamentals and Advances of Orbitrap Mass Spectrometry. In Encyclopedia of Analytical Chemistry; Wiley: Hoboken, NJ, USA, 2019; pp. 1–40. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M.J.N.M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, I.A.; Surin, A.K.; Fornelli, L.; Pridatchenko, M.L.; Suvorina, M.Y.; Gorshkov, M.V. Ion Coalescence in Fourier Transform Mass Spectrometry: Should We Worry about This in Shotgun Proteomics? Eur. J. Mass Spectrom. 2015, 21, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Ledford, E.B.; Rempel, D.L.; Gross, M.L. Space charge effects in Fourier transform mass spectrometry. II. Mass calibration. Anal. Chem. 1984, 56, 2744–2748. [Google Scholar] [CrossRef] [PubMed]
- Pino, L.K.; Just, S.C.; MacCoss, M.J.; Searle, B.C. Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries. J. Mol. Cell. Proteom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Orsburn, B. BoxCar Assisted MS Fragmentation (BAMF). BioRxiv 2019, 860858. [Google Scholar] [CrossRef]
- Erickson, B.K.; Schweppe, D.K.; Yu, Q.; Rad, R.; Haas, W.; McAlister, G.C.; Gygi, S.P. Parallel Notched Gas-Phase Enrichment for Improved Proteome Identification and Quantification with Fast Spectral Acquisition Rates. J. Proteome Res. 2020, 19, 2750–2757. [Google Scholar] [CrossRef]
- Wichmann, C.; Meier, F.; Virreira Winter, S.; Brunner, A.-D.; Cox, J.; Mann, M. MaxQuant.Live Enables Global Targeting of More Than 25,000 Peptides. J. Mol. Cell. Proteom. 2019, 18, 982–994. [Google Scholar] [CrossRef]
- Searle, B.C.; Swearingen, K.E.; Barnes, C.A.; Schmidt, T.; Gessulat, S.; Küster, B.; Wilhelm, M. Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat. Commun. 2020, 11, 1548. [Google Scholar] [CrossRef]
- Hogrebe, A.; von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.T.; Kelstrup, C.D.; Olsen, J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Bouwmeester, R.; Gabriels, R.; Hulstaert, N.; Martens, L.; Degroeve, S. DeepLC can predict retention times for peptides that carry as-yet unseen modifications. BioRxiv 2020. [Google Scholar] [CrossRef]
- Williamson, N.A.; Reilly, C.; Tan, C.T.; Ramarathinam, S.H.; Jones, A.; Hunter, C.L.; Rooney, F.R.; Purcell, A.W. A novel strategy for the targeted analysis of protein and peptide metabolites. Proteomics 2011, 11, 183–192. [Google Scholar] [CrossRef]
- Cumeras, R.; Figueras, E.; Davis, C.E.; Baumbach, J.I.; Gracia, I. Review on ion mobility spectrometry. Part 1: Current instrumentation. Analyst 2015, 140, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Ridgeway, M.E.; Lubeck, M.; Jordens, J.; Mann, M.; Park, M.A. Trapped ion mobility spectrometry: A short review. Int. J. Mass Spectrom. 2018, 425, 22–35. [Google Scholar] [CrossRef]
- Winter, D.L.; Wilkins, M.R.; Donald, W.A. Differential Ion Mobility–Mass Spectrometry for Detailed Analysis of the Proteome. Trends Biotechnol. 2019, 37, 198–213. [Google Scholar] [CrossRef]
- Campuzano, I.D.G.; Giles, K. Historical, current and future developments of travelling wave ion mobility mass spectrometry: A personal perspective. Trac. Trends Anal. Chem. 2019, 120, 115620. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Martínez-Val, A.; Steigerwald, S.; Rüther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef]
- Burger, T. Gentle Introduction to the Statistical Foundations of False Discovery Rate in Quantitative Proteomics. J. Proteome Res. 2018, 17, 12–22. [Google Scholar] [CrossRef]
- Solntsev, S.K.; Shortreed, M.R.; Frey, B.L.; Smith, L.M. Enhanced Global Post-translational Modification Discovery with MetaMorpheus. J. Proteome Res. 2018, 17, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Vitek, O.; Aebersold, R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 2007, 4, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Vitek, O.; Nesvizhskii, A.I. A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinform. 2012, 13, S1. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Yoganathan, S.; Vederas, J.C. 5.02—Nonprotein l-Amino Acids. In Comprehensive Natural Products II; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 5–70. [Google Scholar] [CrossRef]
- Rodgers, K.J.; Samardzic, K.; Main, B.J.J.P.T. Toxic nonprotein amino acids. Plant Toxins 2015, 1, 1–20. [Google Scholar]
- Rodgers, K.J.; Shiozawa, N. Misincorporation of amino acid analogues into proteins by biosynthesis. Int. J. Biochem. Cell Biol. 2008, 40, 1452–1466. [Google Scholar] [CrossRef]
- Torbick, N.; Ziniti, B.; Stommel, E.; Linder, E.; Andrew, A.; Caller, T.; Haney, J.; Bradley, W.; Henegan, P.L.; Shi, X. Assessing Cyanobacterial Harmful Algal Blooms as Risk Factors for Amyotrophic Lateral Sclerosis. Neurotox. Res. 2018, 33, 199–212. [Google Scholar] [CrossRef]
- Silva, D.F.; Candeias, E.; Esteves, A.R.; Magalhães, J.D.; Ferreira, I.L.; Nunes-Costa, D.; Rego, A.C.; Empadinhas, N.; Cardoso, S.M. Microbial BMAA elicits mitochondrial dysfunction, innate immunity activation, and Alzheimer’s disease features in cortical neurons. J. Neuroinflamm. 2020, 17, 332. [Google Scholar] [CrossRef]
- Vega, A. α-Amino-β-methylaminopropionic acid, a new amino acid from seeds of Cycas circinalis. Phytochemistry 1967, 6, 759–762. [Google Scholar] [CrossRef]
- Cox, P.A.; Banack, S.A.; Murch, S.J.; Rasmussen, U.; Tien, G.; Bidigare, R.R.; Metcalf, J.S.; Morrison, L.F.; Codd, G.A.; Bergman, B. Diverse taxa of cyanobacteria produce beta-N-methylamino-L-alanine, a neurotoxic amino acid. Proc. Natl. Acad. Sci. USA 2005, 102, 5074–5078. [Google Scholar] [CrossRef]
- Tomita-Yokotani, K.; Hashimoto, H.; Fujii, Y.; Nakamura, T.; Yamashita, M. Distribution of L-DOPA in the root of velvet bean plant (Mucuna pruriens L.) and gravity. Uchu Seibutsu Kagaku 2004, 18, 165–166. [Google Scholar] [PubMed]
- Ipson, B.R.; Fisher, A.L. Roles of the tyrosine isomers meta-tyrosine and ortho-tyrosine in oxidative stress. Ageing Res. Rev. 2016, 27, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Rehak, L.; Jander, G. meta-Tyrosine in Festuca rubra ssp. commutata (Chewings fescue) is synthesized by hydroxylation of phenylalanine. Phytochemistry 2012, 75, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Constantin, M.-M.; Nita, I.E.; Olteanu, R.; Constantin, T.; Bucur, S.; Matei, C.; Raducan, A. Significance and impact of dietary factors on systemic lupus erythematosus pathogenesis. Exp. Med. 2019, 17, 1085–1090. [Google Scholar] [CrossRef]
- Kitagawa, M.; Tomiyama, T. A new amino-compound in the jack bean and a corresponding new ferment.(I). J. Biochem. 1929, 11, 265–271. [Google Scholar] [CrossRef]
- Rosenthal, G.A. Plant Nonprotein Amino and Imino Acids: Biological, Biochemical, and Toxicological Properties; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Yu, F.; Teo, G.C.; Kong, A.T.; Haynes, S.E.; Avtonomov, D.M.; Geiszler, D.J.; Nesvizhskii, A.I. Identification of modified peptides using localization-aware open search. Nat. Commun. 2020, 11, 4065. [Google Scholar] [CrossRef]
- Chi, H.; Liu, C.; Yang, H.; Zeng, W.F.; Wu, L.; Zhou, W.J.; Wang, R.M.; Niu, X.N.; Ding, Y.H.; Zhang, Y.; et al. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Devabhaktuni, A.; Lin, S.; Zhang, L.; Swaminathan, K.; Gonzalez, C.G.; Olsson, N.; Pearlman, S.M.; Rawson, K.; Elias, J.E. TagGraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nat. Biotechnol. 2019, 37, 469–479. [Google Scholar] [CrossRef]
- Tran, N.H.; Rahman, M.Z.; He, L.; Xin, L.; Shan, B.; Li, M. Complete De Novo Assembly of Monoclonal Antibody Sequences. Sci. Rep. 2016, 6, 31730. [Google Scholar] [CrossRef]
- Li, Q.; Shortreed, M.R.; Wenger, C.D.; Frey, B.L.; Schaffer, L.V.; Scalf, M.; Smith, L.M. Global Post-Translational Modification Discovery. J. Proteome Res. 2017, 16, 1383–1390. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinform. 2012. [Google Scholar] [CrossRef] [PubMed]
- Cvetesic, N.; Semanjski, M.; Soufi, B.; Krug, K.; Gruic-Sovulj, I.; Macek, B. Proteome-wide measurement of non-canonical bacterial mistranslation by quantitative mass spectrometry of protein modifications. Sci. Rep. 2016, 6, 28631. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C.; Pino, L.K.; Egertson, J.D.; Ting, Y.S.; Lawrence, R.T.; MacLean, B.X.; Villen, J.; MacCoss, M.J. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9, 5128. [Google Scholar] [CrossRef] [PubMed]
- Boekel, J.; Chilton, J.M.; Cooke, I.R.; Horvatovich, P.L.; Jagtap, P.D.; Käll, L.; Lehtiö, J.; Lukasse, P.; Moerland, P.D.; Griffin, T.J. Multi-omic data analysis using Galaxy. Nat. Biotechnol. 2015, 33, 137–139. [Google Scholar] [CrossRef]
- Koenig, T.; Menze, B.H.; Kirchner, M.; Monigatti, F.; Parker, K.C.; Patterson, T.; Steen, J.J.; Hamprecht, F.A.; Steen, H. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J. Proteome Res. 2008, 7, 3708–3717. [Google Scholar] [CrossRef]
- Choi, M.; Carver, J.; Chiva, C.; Tzouros, M.; Huang, T.; Tsai, T.H.; Pullman, B.; Bernhardt, O.M.; Huttenhain, R.; Teo, G.C.; et al. MassIVE.quant: A community resource of quantitative mass spectrometry-based proteomics datasets. Nat. Methods 2020. [Google Scholar] [CrossRef]
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Sew, Y.S.; Aizat, W.M.; Razak, M.S.F.A.; Zainal-Abidin, R.-A.; Simoh, S.; Abu-Bakar, N. Comprehensive proteomics data on whole rice grain of selected pigmented and non-pigmented rice varieties using SWATH-MS approach. Data Brief 2020, 31, 105927. [Google Scholar] [CrossRef]
- Rinas, A.; Espino, J.A.; Jones, L.M. An efficient quantitation strategy for hydroxyl radical-mediated protein footprinting using Proteome Discoverer. Anal. Bioanal. Chem. 2016, 408, 3021–3031. [Google Scholar] [CrossRef]
- Gatto, L.; Christoforou, A. Using R and Bioconductor for proteomics data analysis. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 42–51. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Muntel, J.; Gandhi, T.; Verbeke, L.; Bernhardt, O.M.; Treiber, T.; Bruderer, R.; Reiter, L. Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol. Omics 2019, 15, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, O.; Selevsek, N.; Gillet, L.; Rinner, O.; Picotti, P.; Aebersold, R.; Reiter, L. Spectronaut: A Fast and Efficient Algorithm for MRM-Like Processing of Data Independent Acquisition (SWATH-MS) Data. Available online: https://biognosys.com/media.ashx/spectronaut-a-fast-and-efficient-algorithm-for-mrm-like-swath-processing.pdf (accessed on 21 January 2021).
- Shteynberg, D.D.; Deutsch, E.W.; Campbell, D.S.; Hoopmann, M.R.; Kusebauch, U.; Lee, D.; Mendoza, L.; Midha, M.K.; Sun, Z.; Whetton, A.D.; et al. PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res. 2019, 18, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Analysis Type | Method of Analysis | Ref. |
|---|---|---|
| Direct | -MS mass shift analysis | [57] |
| -MS/MS peptide spectral analysis | NPD | |
| -MS-READ | [25] | |
| Indirect | -Hydrolysed amino acid analysis | [41] |
| -Radio-labelled amino acid analysis | [40] | |
| -Amino acid competition studies | [32] | |
| -tRNA micro-assay | [40] | |
| -Proteoform isoelectric point analysis | [47] | |
| -Enzymatic activity assays | [48] | |
| -Dinitrophenyl assay | [36] |
| Site of Modification | Letter Symbol | Modification |
|---|---|---|
| Alanine | A | Carbonylation |
| Arginine | R | Hydroxylation, Phosphorylation, Methylation, ADP-ribosylation, Citrullination, Carbonylation |
| Asparagine | N | Hydroxylation, Methylation, N-linked glycosylation |
| Aspartic acid | D | Hydroxylation, Phosphorylation, Methylation |
| Cysteine | C | Hydroxylation, Phosphorylation, Methylation, Sulfation, Myristoylation, ADP-ribosylation, Nitrosylation |
| Glutamic acid | E | Phosphorylation, Methylation, ADP-ribosylation |
| Glutamine | Q | Methylation, Carbonylation |
| Glycine | G | Myristoylation |
| Histidine | H | Phosphorylation, Methylation |
| Isoleucine | I | Methylation, Carbonylation |
| Leucine | L | Methylation |
| Lysine | K | Hydroxylation, Phosphorylation, Methylation, Ubiquitination, Myristoylation, ADP-ribosylation, Carbonylation, Malonylation, Succinylation, Glutarylation, Biotinylation |
| Methionine | M | Hydroxylation |
| Phenylalanine | F | Hydroxylation |
| Proline | P | Hydroxylation |
| Serine | S | Phosphorylation, Methylation, Sulfation, O-linked glycosylation, Carbonylation, Decanoylation |
| Selenocysteine | U | Hydroxylation |
| Threonine | T | Phosphorylation, Methylation, Sulfation, O-linked glycosylation, Decanoylation |
| Tryptophan | W | Glycosylation, Bromination, Quinone |
| Tyrosine | Y | Hydroxylation, Phosphorylation, Sulfation, O-linked glycosylation, Quinone |
| Valine | V | Hydroxylation, Carbonylation |
| NPAA | Homologue | Mass Shift | Theoretical Immonium Ion (m/z) | Disease Associated with Toxicity | Source |
|---|---|---|---|---|---|
| BMAA | Serine | +13.0316 | 73.0766 | MND [99] AD/PD [100] | Cycad palms and cyanobacteria [101,102] |
| Alanine | +29.0266 | ||||
| L-DOPA * | Tyrosine | +15.9949 | 152.0712 | PD [15] | Velvet bean plant (Mucuna pruriens) [103] |
| Phenylalanine | +31.9898 | ||||
| Meta-tyrosine * | Tyrosine | NMS | 136.0762 | AD/PD [104] | Fescue grass (Festuca spp.) [105] |
| Phenylalanine | +15.9949 | ||||
| Ortho-tyrosine * | Tyrosine | NMS | 136.0762 | AD/PD, marker of Asctheleroisis [104] | Oxidation product of phenylalanine |
| Phenylalanine | +15.9949 | ||||
| Norvaline | Leucine | −14.0157 | 72.08132 | Cytotoxic [61] | Nutritional supplement [61] |
| Isoleucine | |||||
| Valine | NMS | ||||
| Azetidine 2 carboxylic acid | Proline | −14.0157 | 56.05002 | MS [19] | Lily of the valley (Convallaria majalis); |
| Garden beet (Beta vulgaris) [9] | |||||
| Canavanine | Arginine | +1.9793 | 131.0933 | MS/Systemic lupus erythematosus [106] | Jack bean plant (Canavalia ensiformis) [107] |
| Norleucine | Methionine | −17.9564 | 86.09697 | NNAD | Bacteria |
| Mimosine | Tyrosine | +16.9902 | 153.0664 | NNAD | Leucaena (Leucaena spp.) and some Mimosa species [108] |
| Program | GUI | Cost | Open Searches | Accessibility | DIA Searching | Paper |
|---|---|---|---|---|---|---|
| Byonic | Yes | Licensed | Yes | Easy | No | [114] |
| EncyclopeDIA | Yes | Free | No | Easy | Exclusively | [116] |
| Fragpipe | Yes | Free to academics | Yes | Easy | Yes | [109] |
| Msfragger, Philosopher, PTMShepard | ||||||
| Galaxy P | Yes | Free | No | Intermediary | Being implemented | [117] |
| Mascot | Yes | Licensed | No | Easy | No | [118] |
| MassIVE | Yes | Free | Yes | Easy | Yes | [119] |
| Maxquant | Yes | Free | Yes, Dependent peptides | Intermediary | No | [92] |
| Andromeda | ||||||
| MetaMorpheus | Yes | Free | Yes | Intermediary | No | [91] |
| OpenMS | Yes | Free | Yes * | Advanced | Yes | [120] |
| Open-pFind | Yes | Free Licensed | Yes | Easy | No | [110] |
| Peaks Studio | Yes | Licensed | Yes | Easy | Yes | [121] |
| Protein Pilot, PeakView | Yes | Licensed | Yes: Protein Pilot | Easy | Yes: PeakView | [122] |
| Proteome Discoverer | Yes | Licensed | Through Nodes | Easy | Yes | [123] |
| R workflows * | Yes | Free | Yes | Advanced | Yes | [124] |
| Skyline | Yes | Free | N/A | Intermediary | No | [125] |
| Signal quantification (DIA, MRM, PRM) | ||||||
| SpectroMine | Yes | Licensed | Yes | Easy | No | [126] |
| PTM Shepard | ||||||
| Spectronaut | Yes | Licensed | No | Easy | Yes | [127] |
| TagGraph | No | Free | Yes | Advanced | No | [111] |
| Trans-Proteomic Pipeline | Yes | Free | Yes | Advanced | No | [128] |
| PTMProphet |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steele, J.R.; Italiano, C.J.; Phillips, C.R.; Violi, J.P.; Pu, L.; Rodgers, K.J.; Padula, M.P. Misincorporation Proteomics Technologies: A Review. Proteomes 2021, 9, 2. https://doi.org/10.3390/proteomes9010002
Steele JR, Italiano CJ, Phillips CR, Violi JP, Pu L, Rodgers KJ, Padula MP. Misincorporation Proteomics Technologies: A Review. Proteomes. 2021; 9(1):2. https://doi.org/10.3390/proteomes9010002
Chicago/Turabian StyleSteele, Joel R., Carly J. Italiano, Connor R. Phillips, Jake P. Violi, Lisa Pu, Kenneth J. Rodgers, and Matthew P. Padula. 2021. "Misincorporation Proteomics Technologies: A Review" Proteomes 9, no. 1: 2. https://doi.org/10.3390/proteomes9010002
APA StyleSteele, J. R., Italiano, C. J., Phillips, C. R., Violi, J. P., Pu, L., Rodgers, K. J., & Padula, M. P. (2021). Misincorporation Proteomics Technologies: A Review. Proteomes, 9(1), 2. https://doi.org/10.3390/proteomes9010002