Comparative Proteome-Wide Analysis of Bone Marrow Microenvironment of β-Thalassemia/Hemoglobin E

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bone Marrow Samples
2.2. Protein Fractionation by One-Dimensional SDS-PAGE
2.3. In-Gel Digestion of Proteins for LC-MS/MS
2.4. LC-MS/MS Analysis
2.5. Database Searching and Protein Quantitation and Identification
2.6. Bioinformatic Analysis
2.7. Validation of Candidate Protein Expression Using Western Blot
2.8. Statistical Evaluation
3. Results
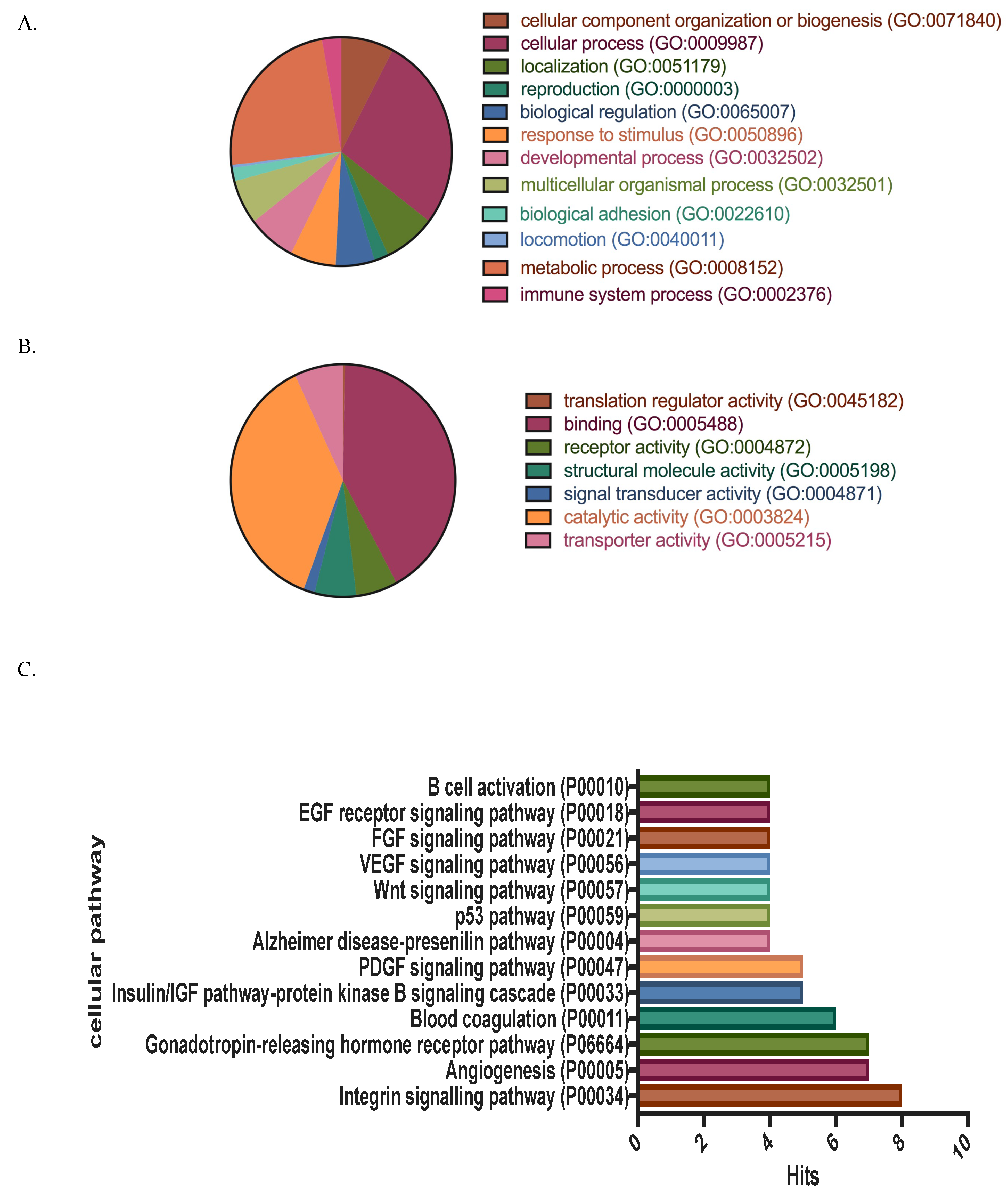
3.1. Protein Identification, Classification and Categorization of the Bone Marrow Microenvironment Protein Profile from β-Thalassemia/Hb E
3.2. ApoD Protein and Validation of the Proteome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| β-thalassemia/HbE | beta thalassemia with variant hemoglobin E |
| ESI-QTOF-MS/MS | electrospray-ionisation-quadrupole-time-of-flight tandem mass spectrometry |
| STRING | Search Tool for the Retrieval of Interacting Genes |
| PPIs | protein-protein interaction networks |
| APOD | Apolipoprotein D |
| HSCs | hematopoietic stem cells |
References
- Rund, D.; Rachmilewitz, E. β-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Rund, D. Thalassemia 2016: Modern medicine battles an ancient disease. Am. J. Hematol. 2016, 91, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Arlet, J.B.; Dussiot, M.; Moura, I.C.; Courtois, G.; Hermine, O. Ineffective erythropoiesis in β-thalassemia. Sci. World J. 2013, 2013, 394295. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. Thalassemia as a global health problem: recent progress toward its control in the developing countries. Ann. N. Y. Acad. Sci. 2010, 1202, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, Y.; Rivella, S. β-thalassemia: A model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011, 118, 4321–4330. [Google Scholar] [CrossRef] [PubMed]
- Gardenghi, S.; Grady, R.W.; Rivella, S. Anemia, ineffective erythropoiesis, and hepcidin: Interacting factors in abnormal iron metabolism leading to iron overload in β-thalassemia. Hematol. Oncol. Clin. North Am. 2010, 24, 1089–1107. [Google Scholar] [CrossRef] [PubMed]
- Mathias, L.A.; Fisher, T.C.; Zeng, L.; Meiselman, H.J.; Weinberg, K.I.; Hiti, A.L.; Malik, P. Ineffective erythropoiesis in β-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp. Hematol. 2000, 28, 1343–1353. [Google Scholar] [CrossRef]
- Pootrakul, P.; Sirankapracha, P.; Hemsorach, S.; Moungsub, W.; Kumbunlue, R.; Piangitjagum, A.; Wasi, P.; Ma, L.; Schrier, S.L. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood 2000, 96, 2606–2612. [Google Scholar] [PubMed]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in β-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Rivella, S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012, 26 (Suppl. 1), S12–S15. [Google Scholar] [CrossRef]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Franco, S.S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and β-thalassemic erythroid cells behind the molecular defect. Oxid. Med. Cell Longev. 2013, 2013, 985210. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, R.E.; Sibmooh, N.; Fucharoen, S.; Friedman, J.M. HbE/β-Thalassemia and Oxidative Stress: The Key to Pathophysiological Mechanisms and Novel Therapeutics. Antioxid. Redox Signal. 2017, 26, 794–813. [Google Scholar] [CrossRef] [PubMed]
- Ponnikorn, S.; Panichakul, T.; Sresanga, K.; Wongborisuth, C.; Roytrakul, S.; Hongeng, S.; Tungpradabkul, S. Phosphoproteomic analysis of apoptotic hematopoietic stem cells from hemoglobin E/β-thalassemia. J. Transl. Med. 2011, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Nwajei, F.; Konopleva, M. The bone marrow microenvironment as niche retreats for hematopoietic and leukemic stem cells. Adv. Hematol. 2013, 2013, 953982. [Google Scholar] [CrossRef] [PubMed]
- Kaur-Bollinger, P.; Gotze, K.S.; Oostendorp, R.A. Role of secreted factors in the regulation of hematopoietic stem cells by the bone marrow microenvironment. Front Biosci. (Landmark Ed.) 2012, 17, 876–891. [Google Scholar] [CrossRef] [PubMed]
- Carrancio, S.; Blanco, B.; Romo, C.; Muntion, S.; Lopez-Holgado, N.; Blanco, J.F.; Briñon, J.G.; San Miguel, J.F.; Sanchez-Guijo, F.M.; Del Cañizo, M.C. Bone marrow mesenchymal stem cells for improving hematopoietic function: An in vitro and in vivo model. Part 2: Effect on bone marrow microenvironment. PLoS ONE 2011, 6, e26241. [Google Scholar] [CrossRef] [PubMed]
- Lithanatudom, P.; Smith, D.R. Analysis of protein profiling studies of β-thalassemia/Hb E disease. Proteomics Clin. Appl. 2016, 10, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Wikan, N.; Ubol, S.; Auewarakul, P.; Paemanee, A.; Kittisenachai, S.; Roytrakul, S.; Smith, D.R. Proteomic analysis of chikungunya virus infected microgial cells. PLoS ONE 2012, 7, e34800. [Google Scholar] [CrossRef] [PubMed]
- Kurdrid, P.; Senachak, J.; Sirijuntarut, M.; Yutthanasirikul, R.; Phuengcharoen, P.; Jeamton, W.; Roytrakul, S.; Cheevadhanarak, S.; Hongsthong, A. Comparative analysis of the Spirulina platensis subcellular proteome in response to low- and high-temperature stresses: uncovering cross-talk of signaling components. Proteome Sci. 2011, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Panichakul, T.; Ponnikorn, S.; Roytrakul, S.; Paemanee, A.; Kittisenachai, S.; Hongeng, S.; Udomsangpetch, R. Plasmodium vivax inhibits erythroid cell growth through altered phosphorylation of the cytoskeletal protein ezrin. Malar J. 2015, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Terrisse, L.; Marcoux, K.; Do Carmo, S.; Brissette, L.; Milne, R.; Rassart, E. Structure-function relationships of human apolipoprotein D an immunochemical analysis. Life Sci. 2001, 70, 629–638. [Google Scholar] [CrossRef]
- Rassart, E.; Bedirian, A.; Do Carmo, S.; Guinard, O.; Sirois, J.; Terrisse, L.; Milne, R. Apolipoprotein D. Biochim. Biophys Acta. 2000, 1482, 185–198. [Google Scholar] [CrossRef]
- Perdomo, G.; Henry Dong, H. Apolipoprotein D in lipid metabolism and its functional implication in atherosclerosis and aging. Aging (Albany NY). 2009, 1, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Soiland, H.; Soreide, K.; Janssen, E.A.; Korner, H.; Baak, J.P.; Soreide, J.A. Emerging concepts of apolipoprotein D with possible implications for breast cancer. Cell Oncol. 2007, 29, 195–209. [Google Scholar] [PubMed]
- Do Carmo, S.; Levros, L.C.; Rassart, E., Jr. Modulation of apolipoprotein D expression and translocation under specific stress conditions. Biochim. Biophys. Acta 2007, 1773, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Leecharoenkiat, A.; Wannatung, T.; Lithanatudom, P.; Svasti, S.; Fucharoen, S.; Chokchaichamnankit, D.; Srisomsap, C.; Smith, D.R. Increased oxidative metabolism is associated with erythroid precursor expansion in β0-thalassaemia/Hb E disease. Blood Cells Mol. Dis. 2011, 47, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Najyb, O.; Do Carmo, S.; Alikashani, A.; Rassart, E. Apolipoprotein D Overexpression Protects Against Kainate-Induced Neurotoxicity in Mice. Mol. Neurobiol. 2016, 54, 3948–3963. [Google Scholar] [CrossRef] [PubMed]
- Bajo-Graneras, R.; Sanchez, D.; Gutierrez, G.; Gonzalez, C.; Do Carmo, S.; Rassart, E.; Ganfornina, M.D. Apolipoprotein D alters the early transcriptional response to oxidative stress in the adult cerebellum. J. Neurochem. 2011, 117, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Ganfornina, M.D.; Do Carmo, S.; Lora, J.M.; Torres-Schumann, S.; Vogel, M.; Allhorn, M.; González, C.; Bastiani, M.J.; Rassart, E.; Sanchez, D. Apolipoprotein D is involved in the mechanisms regulating protection from oxidative stress. Aging Cell. 2008, 7, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Waldner, A.; Dassati, S.; Redl, B.; Smania, N.; Gandolfi, M. Apolipoprotein D Concentration in Human Plasma during Aging and in Parkinson’s Disease: A Cross-Sectional Study. Parkinson’s Dis. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Provost, P.R.; Villeneuve, L.; Weech, P.K.; Milne, R.W.; Marcel, Y.L.; Rassart, E. Localization of the major sites of rabbit apolipoprotein D gene transcription by in situ hybridization. J. Lipid Res. 1991, 32, 1959–1970. [Google Scholar] [PubMed]
- Unchern, S.; Laohareungpanya, N.; Sanvarinda, Y.; Pattanapanyasat, K.; Tanratana, P.; Chantharaksri, U.; Sibmooh, N. Oxidative modification and poor protective activity of HDL on LDL oxidation in thalassemia. Lipids 2010, 45, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Ghorban, K.; Shanaki, M.; Mobarra, N.; Azad, M.; Asadi, J.; Pakzad, R.; Ehteram, H. Apolipoproteins A1, B, and other prognostic biochemical cardiovascular risk factors in patients with β-thalassemia major. Hematology 2016, 21, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Alaupovic, P.; Schaefer, E.J.; McConathy, W.J.; Fesmire, J.D.; Brewer, H.B., Jr. Plasma apolipoprotein concentrations in familial apolipoprotein A-I and A-II deficiency (Tangier disease). Metabolism 1981, 30, 805–809. [Google Scholar] [CrossRef]
- Levros, L.C., Jr.; Do Carmo, S.; Edouard, E.; Legault, P.; Charfi, C.; Rassart, E. Characterization of nuclear factors modulating the apolipoprotein D promoter during growth arrest: Implication of PARP-1, APEX-1 and ERK1/2 catalytic activities. Biochim. Biophys. Acta 2010, 1803, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Wannatung, T.; Lithanatudom, P.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Increased erythropoiesis of β-thalassaemia/Hb E proerythroblasts is mediated by high basal levels of ERK1/2 activation. Br. J. Haematol. 2009, 146, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Sarjeant, J.M.; Lawrie, A.; Kinnear, C.; Yablonsky, S.; Leung, W.; Massaeli, H.; Prichett, W.; Veinot, J.P.; Rassart, E.; Rabinovitch, M. Apolipoprotein D inhibits platelet-derived growth factor-BB-induced vascular smooth muscle cell proliferated by preventing translocation of phosphorylated extracellular signal regulated kinase 1/2 to the nucleus. Arterioscler Thromb Vasc Biol. 2003, 23, 2172–2177. [Google Scholar] [CrossRef] [PubMed]
- Thanuthanakhun, N.; Nuntakarn, L.; Sampattavanich, S.; Anurathapan, U.; Phuphanitcharoenkun, S.; Pornpaiboonstid, S.; Borwornpinyo, S.; Hongeng, S. Investigation of FoxO3 dynamics during erythroblast development in β-thalassemia major. PLoS ONE 2017, 12, e0187610. [Google Scholar] [CrossRef] [PubMed]
- Tia, N.; Singh, A.K.; Pandey, P.; Azad, C.S.; Chaudhary, P.; Gambhir, I.S. Role of Forkhead Box O (FOXO) transcription factor in aging and diseases. Gene 2018, 648, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hull-Thompson, J.; Muffat, J.; Sanchez, D.; Walker, D.W.; Benzer, S.; Ganfornina, M.D.; Jasper, H. Control of metabolic homeostasis by stress signaling is mediated by the lipocalin NLaz. PLoS Genet. 2009, 5, e1000460. [Google Scholar] [CrossRef] [PubMed]
- Pajaniappan, M.; Glober, N.K.; Kennard, S.; Liu, H.; Zhao, N.; Lilly, B. Endothelial cells downregulate apolipoprotein D expression in mural cells through paracrine secretion and Notch signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H784–H793. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot | Name | Relative Protein Expression * | KEGG Analysis |
|---|---|---|---|
| ACLY_HUMAN | ATP citrate lyase | over expression # | TCA cycle |
| APOD_HUMAN | apolipoprotein D | under expression # | Exosomal proteins of other cancer cells |
| ATD3A_HUMAN | ATPase family AAA domain-containing protein 3A | under expression# | Mitochondrial biogenesis |
| BHA09_HUMAN | Class A basic helix-loop-helix protein | 0.785 | - |
| CTCFL_HUMAN | Transcriptional repressor CTCFL | under expression # | - |
| CTNB1_HUMAN | Catenin beta-1 | under expression # | 1 Wnt signaling pathway |
| 2 Hippo signaling pathway | |||
| 3 Signaling pathways regulating pluripotency of stem cells | |||
| DCA15_HUMAN | DDB1- and CUL4-associated factor 15 | 1.368 | Ubiquitin system |
| RPOM_HUMAN | DNA-directed RNA polymerase, mitochondrial | over expression # | Mitochondrial biogenesis |
| TRFE_HUMAN | Serotransferrin | under expression# | 1 HIF-1 signaling pathway |
| 2 Ferroptosis | |||
| 3 Mineral absorption | |||
| TTHY_HUMAN | Transthyretin | 1.002 | Thyroid hormone synthesis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponnikorn, S.; Mongkolrob, R.; Klongthalay, S.; Roytrakul, S.; Srisanga, K.; Tungpradabkul, S.; Hongeng, S. Comparative Proteome-Wide Analysis of Bone Marrow Microenvironment of β-Thalassemia/Hemoglobin E. Proteomes 2019, 7, 8. https://doi.org/10.3390/proteomes7010008
Ponnikorn S, Mongkolrob R, Klongthalay S, Roytrakul S, Srisanga K, Tungpradabkul S, Hongeng S. Comparative Proteome-Wide Analysis of Bone Marrow Microenvironment of β-Thalassemia/Hemoglobin E. Proteomes. 2019; 7(1):8. https://doi.org/10.3390/proteomes7010008
Chicago/Turabian StylePonnikorn, Saranyoo, Rungrawee Mongkolrob, Suwit Klongthalay, Sittiruk Roytrakul, Kitima Srisanga, Sumalee Tungpradabkul, and Suradej Hongeng. 2019. "Comparative Proteome-Wide Analysis of Bone Marrow Microenvironment of β-Thalassemia/Hemoglobin E" Proteomes 7, no. 1: 8. https://doi.org/10.3390/proteomes7010008
APA StylePonnikorn, S., Mongkolrob, R., Klongthalay, S., Roytrakul, S., Srisanga, K., Tungpradabkul, S., & Hongeng, S. (2019). Comparative Proteome-Wide Analysis of Bone Marrow Microenvironment of β-Thalassemia/Hemoglobin E. Proteomes, 7(1), 8. https://doi.org/10.3390/proteomes7010008