
Towards the Full Realization of 2DE Power

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Power of 2DE Separation
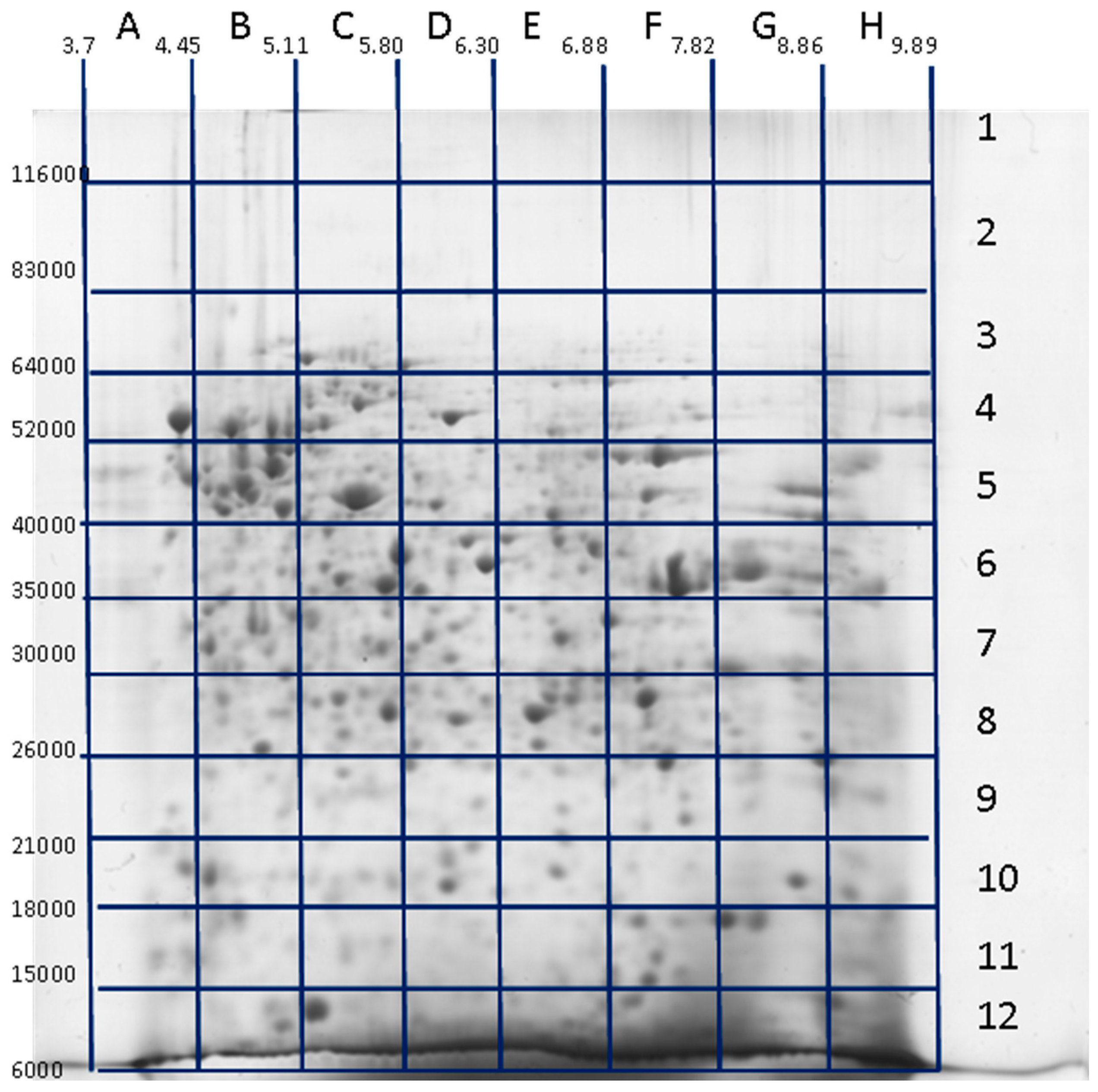
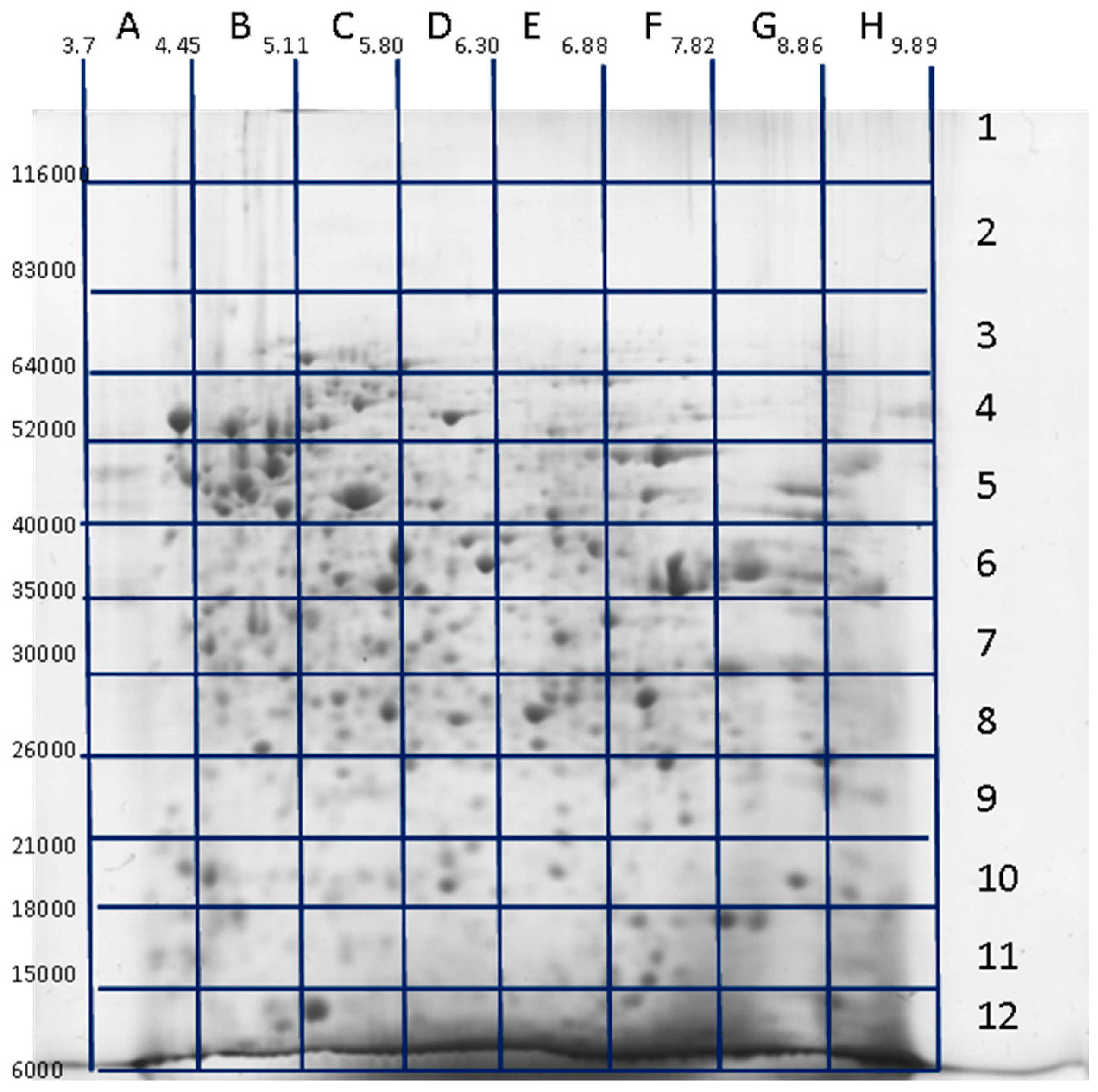
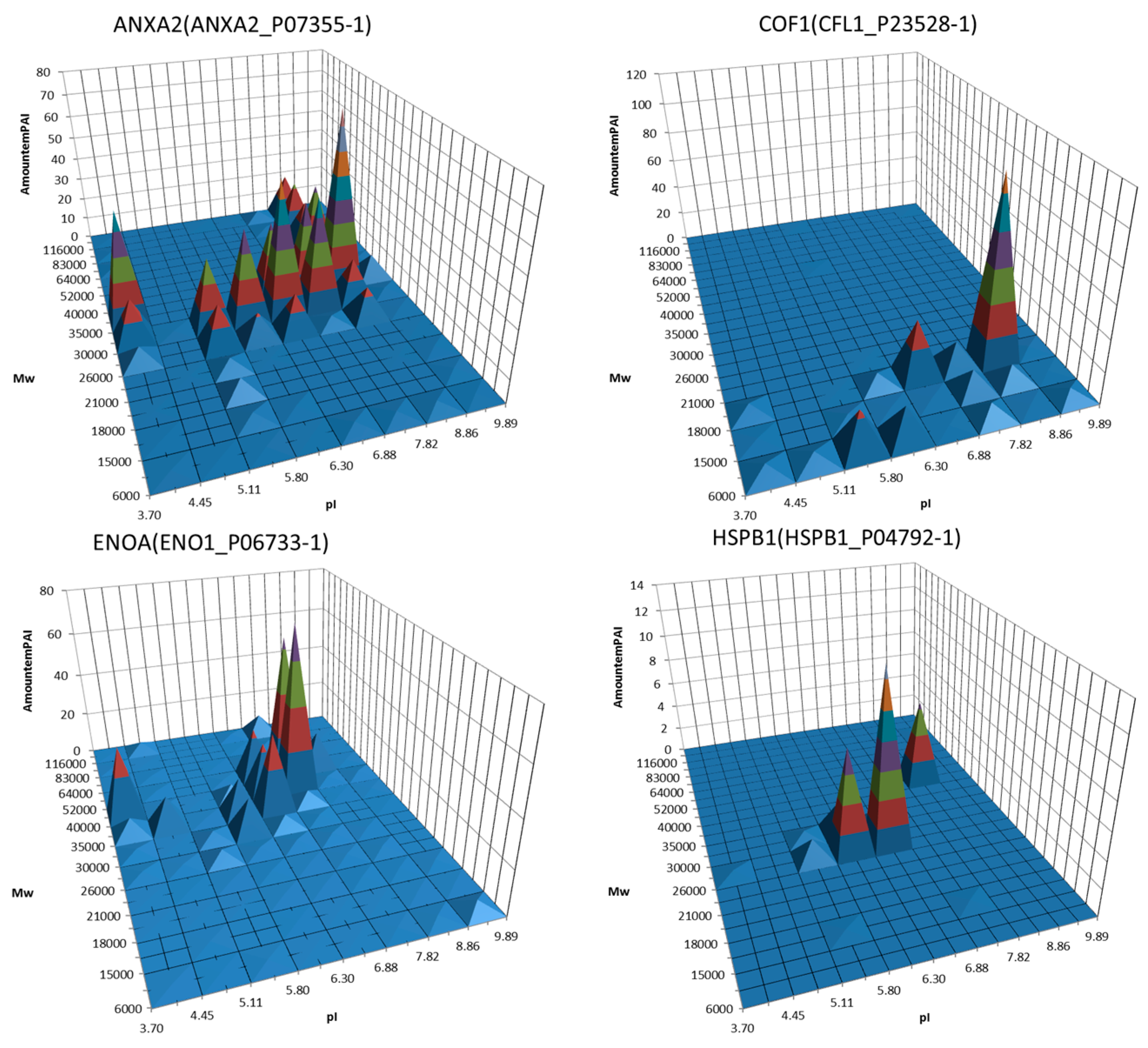
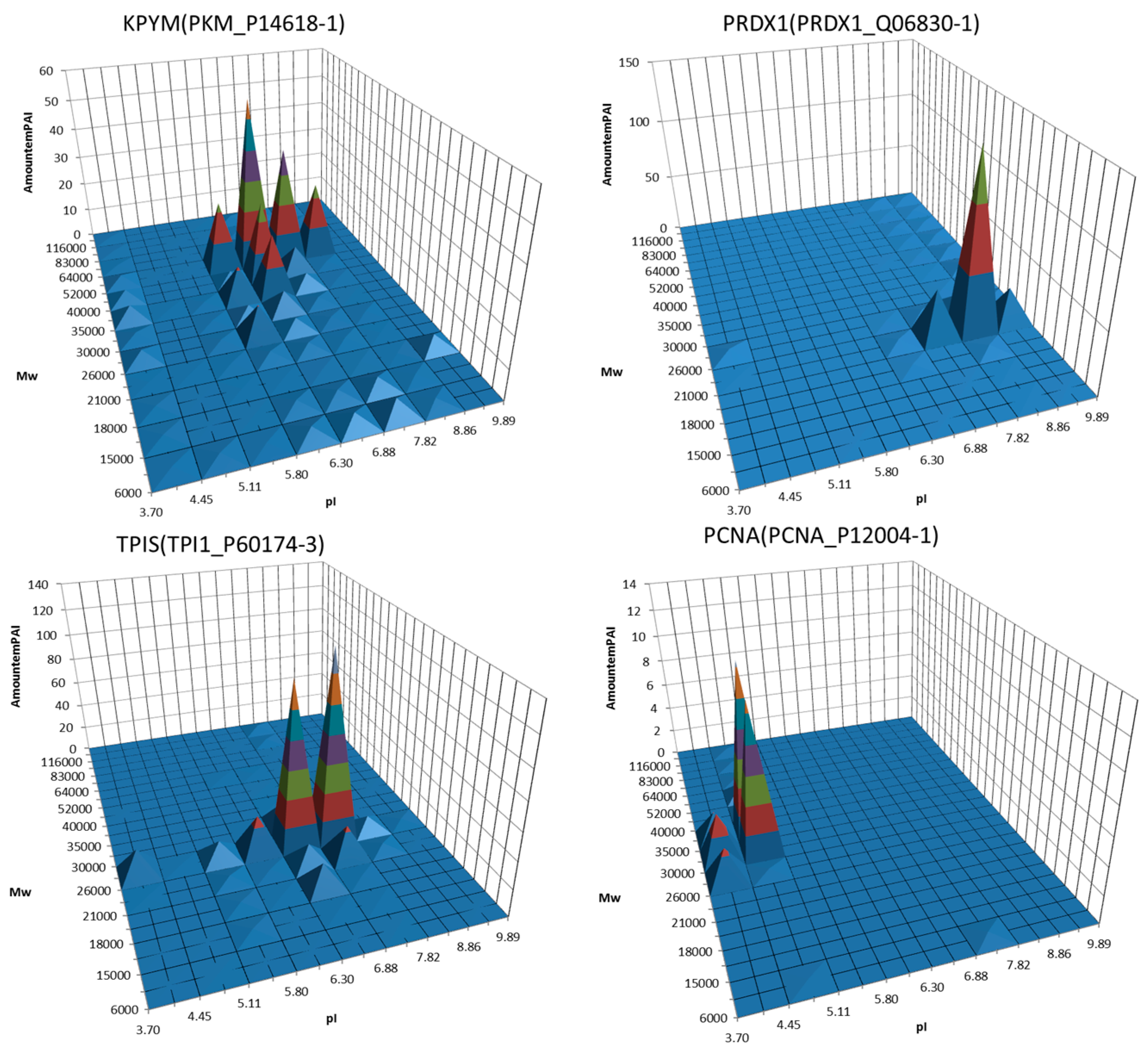
3. Decoding of Information Hidden inside the 2DE Gel
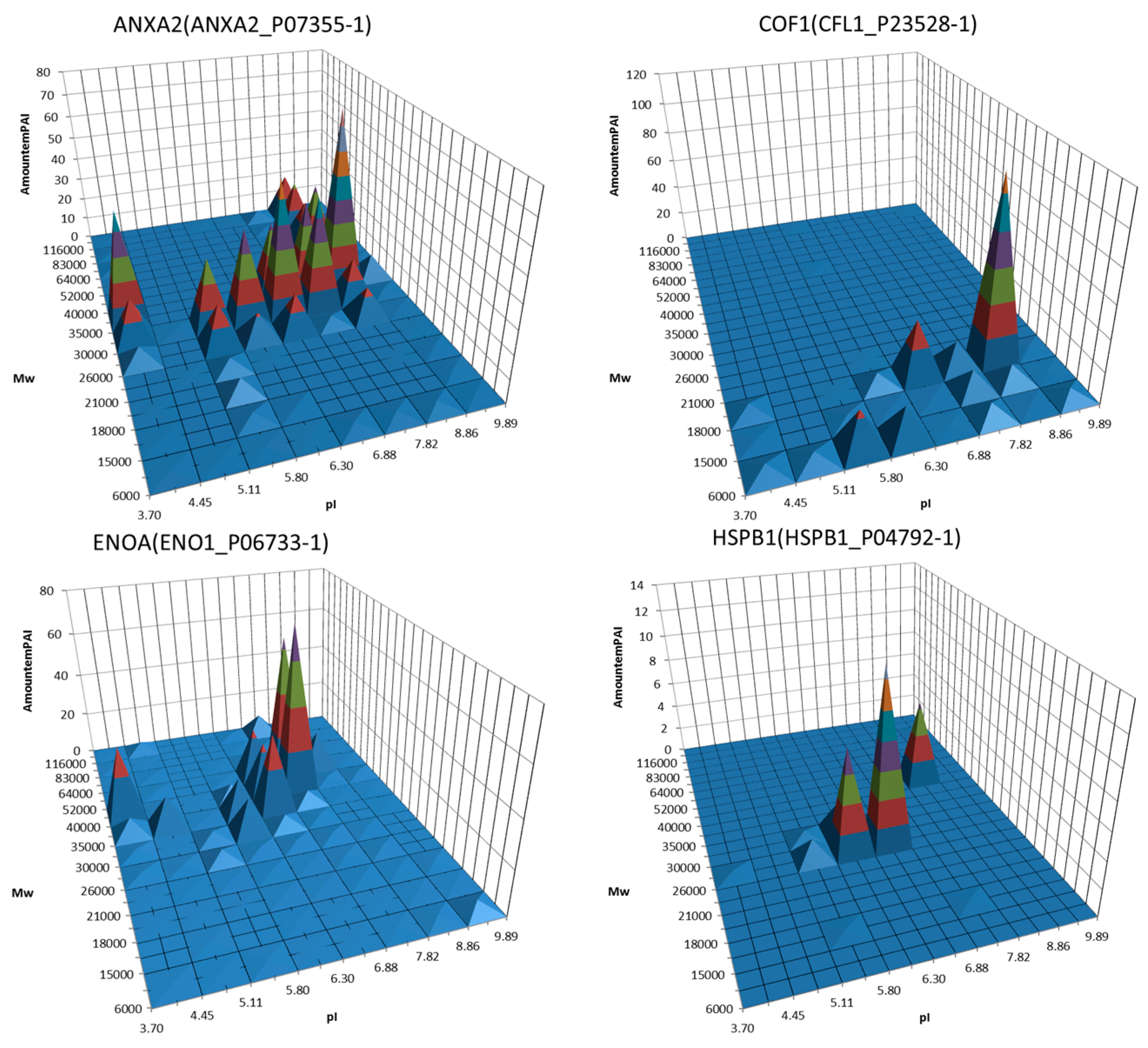
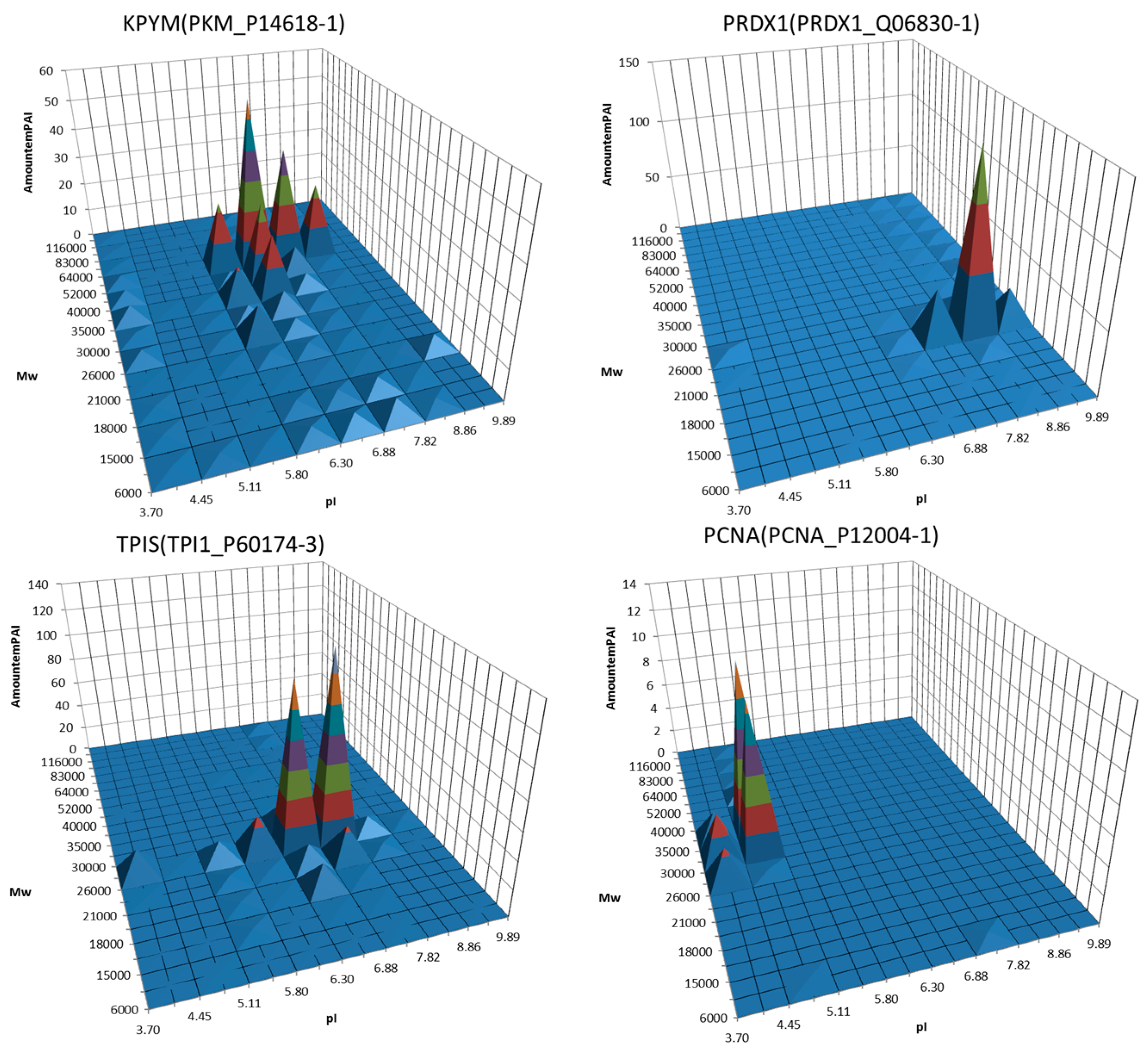
4. Utility of 2DE Separation Parameters
5. 2DE Databases
6. Virtual 2DE
7. Virtual-Experimental 2DE
8. Conclusions
- (1)
- sample preparation/extraction
- (2)
- protein separation by 2DE
- (3)
- protein gel staining (Coomassie)
- (4)
- proteoform location, identification, characterization, and quantitation by ESI LC-MS/MS
- (5)
- virtual/experimental 2DE protein database construction
Acknowledgments
Conflicts of Interest
References
- Anderson, N.G.; Anderson, L. The Human Protein Index. Clin. Chem. 1982, 28, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Anderson, N.G. High resolution two-dimensional electrophoresis of human plasma proteins. Proc. Natl. Acad. Sci. USA 1977, 74, 5421–5425. [Google Scholar] [CrossRef]
- Anderson, N.G.; Matheson, A.; Anderson, N.L. Back to the future: The human protein index (HPI) and the agenda for post-proteomic biology. Proteomics 2001, 1, 3–12. [Google Scholar] [CrossRef]
- Wilkins, M. Proteomics data mining. Expert Rev. Proteom. 2009, 6, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Jungblut, P.R. The proteomics quantification dilemma. J. Proteom. 2014, 107, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Jungblut, P.R. Back to the future—The value of single protein species investigations. Proteomics 2013, 13, 3103–3105. [Google Scholar] [CrossRef] [PubMed]
- Jungblut, P.R.; Schlüter, H. Towards the analysis of protein species: An overview about strategies and methods. Amino Acids 2011, 41, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jensen, O.N. Modification-specific proteomics: Strategies for characterization of post-translational modifications using enrichment techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef] [PubMed]
- Klose, J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik 1975, 26, 231–243. [Google Scholar] [PubMed]
- Klose, J. From 2-D electrophoresis to proteomics. Electrophoresis 2009, 30, 142–149. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Scheele, G.A. Two-dimensional gel analysis of soluble proteins. Charaterization of guinea pig exocrine pancreatic proteins. J. Biol. Chem. 1975, 250, 5375–5385. [Google Scholar] [PubMed]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A.; Makarov, A. Orbitrap mass spectrometry. Anal. Chem. 2013, 85, 5288–5296. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.C.; Edwards, A.V.G.; Verano-Braga, T.; Schwämmle, V.; Kjeldsen, F.; Jensen, O.N.; Larsen, M.R. High-performance hybrid Orbitrap mass spectrometers for quantitative proteome analysis: Observations and implications. Proteomics 2016, 16, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R. Mass spectral analysis in proteomics. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, G.; Turck, C.W.; Martins-De-souza, D. Shotgun mass spectrometry workflow combining IEF and LC-MALDI-TOF/TOF. Protein J. 2010, 29, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Primo, V.; Smejkal, G.B.; Moskovets, E.V.; Kuo, W.P.; Ivanov, A.R. Comparison of in-gel protein separation techniques commonly used for fractionation in mass spectrometry-based proteomic profiling. Electrophoresis 2012, 33, 2516–2526. [Google Scholar] [CrossRef] [PubMed]
- Giorgianni, F.; Desiderio, D.M.; Beranova-giorgianni, S.; Neuroscience, C.B.S. Proteome analysis using isoelectric focusing in immobilized pH gradient gels followed by mass spectrometry. Electrophoresis 2003, 24, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.-J.; Dai, J.; Li, S.-J.; Sheng, Q.-H.; Deng, W.-J.; Xia, Q.-C.; Zeng, R. High-sensitivity analysis of human plasma proteome by immobilized isoelectric focusing fractionation coupled to mass spectrometry identification. J. Proteome Res. 2005, 4, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Lisitsa, A.V.; Petushkova, N.A.; Thiele, H.; Moshkovskii, S.A.; Zgoda, V.G.; Karuzina, I.I.; Chernobrovkin, A.L.; Skipenko, O.G.; Archakov, A.I. Application of slicing of one-dimensional gels with subsequent slice-by-slice mass spectrometry for the proteomic profiling of human liver cytochromes P450. J. Proteome Res. 2010, 9, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Spicer, V.; Ezzati, P.; Neustaeter, H.; Beavis, R.C.; Wilkins, J.A.; Krokhin, O.V. 3D HPLC-MS with Reversed-Phase Separation Functionality in All Three Dimensions for Large-Scale Bottom-Up Proteomics and Peptide Retention Data Collection. Anal. Chem. 2016, 88, 2847–2855. [Google Scholar] [CrossRef] [PubMed]
- Smithies, O.; Poulik, M.D. Two-dimensional electrophoresis of serum proteins. Nature 1956, 177, 1033. [Google Scholar] [CrossRef] [PubMed]
- Bussard, A.; Huet, J.M. Description of a technic simultaneously combining electrophoresis and immunological precipitation in gel: Electrosyneresis. Biochim. Biophys. Acta 1959, 34, 258–260. [Google Scholar] [CrossRef]
- Laurell, C.B. Electroimmuno Assay. Scand. J. Clin. Lab. Investig. 1972, 29, 21–37. [Google Scholar] [CrossRef]
- Laurell, C.-B. Quantitative estimation of proteins by electrophoresis in agarose gel containing antibodies. Anal. Biochem. 1966, 15, 45–52. [Google Scholar] [CrossRef]
- Raymond, S.; Weintraub, L. Acrylamide gel as a supporting medium for zone electrophoresis. Science 1959, 130, 711. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, E.; Wittmann, H.G. Ribosomal proteins. VII. Two-dimensional polyacrylamide gel electrophoresis for fingerprinting of ribosomal proteins. Anal. Biochem. 1970, 36, 401–412. [Google Scholar] [CrossRef]
- Awdeh, Z.L.; Williamson, A.R.; Askonas, B.A. Isoelectric focusing in polyacrylamide gels. Nature 1968, 219, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G.; Drysdale, J.W. Isoelectric focusing in gels. J. Chromatogr. A 1974, 98, 271–321. [Google Scholar] [CrossRef]
- Dale, G.; Latner, A.L. Isoelectric focusing in polyacrylamide gels. Lancet 1968, 1, 847–848. [Google Scholar] [CrossRef]
- Fawcett, J.S. Isoelectric fractionation of proteins on polyacrylamide gels. FEBS Lett. 1968, 1, 81–82. [Google Scholar] [CrossRef]
- Ornstein, L. Disc Electrophoresis. I. Background and Theory. Ann. N. Y. Acad. Sci. 1964, 121, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J. Disc Electrophoresis—II Method and Application to Human Serum Proteins. Ann. N. Y. Acad. Sci. 1964, 121, 404–427. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, M.; Righetti, P.G. Proteomics Today: Protein Assessment and Biomarkers Using Mass Spectrometry, 2D Electrophoresis, and Microarray Technology; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Gorg, A.; Obermaier, C.; Boguth, G.; Harder, A.; Scheibe, B.; Wildgruber, R.; Weiss, W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 2000, 21, 1037–1053. [Google Scholar] [CrossRef]
- Gorg, A.; Postel, W.; Domscheit, A.; Gunther, S. Two-dimensional electrophoresis with immobilized pH gradients of leaf proteins from barley (Hordeum vulgare): Method, reproducibility and genetic aspects. Electrophoresis 1988, 9, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G. Recent developments in electrophoretic methods. J. Chromatogr. A 1990, 516, 3–22. [Google Scholar] [CrossRef]
- Vecchio, G.; Righetti, P.G.; Zanoni, M.; Artoni, G.; Gianazza, E. Fractionation techniques in a hydro-organic environment. I. Sulfolane as a solvent for hydrophobic proteins. Anal. Biochem. 1984, 137, 410–419. [Google Scholar] [CrossRef]
- Rabilloud, T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis 1998, 19, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Seddon, A.M.; Curnow, P.; Booth, P.J. Membrane proteins, lipids and detergents: Not just a soap opera. Biochim. Biophys. Acta Biomembr. 2004, 1666, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Blisnick, T.; Heller, M.; Luche, S.; Aebersold, R.; Lunardi, J.; Braun-Breton, C. Analysis of membrane proteins by two-dimensional electrophoresis: Comparison of the proteins extracted from normal or Plasmodium falciparum-infected erythrocyte ghosts. Electrophoresis 1999, 20, 3603–3610. [Google Scholar] [CrossRef]
- Smejkal, G.B.; Lazarev, A. Solution phase isoelectric fractionation in the multi-compartment electrolyser: A divide and conquer strategy for the analysis of complex proteomes. Briefings Funct. Genom. Proteom. 2005, 4, 76–81. [Google Scholar] [CrossRef]
- Di Girolamo, F.; Bala, K.; Chung, M.C.M.; Righetti, P.G. “Proteomineering” serum biomarkers. A Study in Scarlet. Electrophoresis 2011, 32, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, E.; Monsarrat, B.; Righetti, P.G. The “ Invisible Proteome “: How to Capture the Low-Abundance Proteins Via Combinatorial Ligand Libraries. Amino Acids 2007, 4, 198–208. [Google Scholar]
- Inagaki, N.; Katsuta, K. Large Gel Two-Dimensional Electrophoresis: Improving Recovery of Cellular Proteome. Curr. Proteom. 2004, 1, 35–39. [Google Scholar] [CrossRef]
- Zuo, X.U.N.; Lee, K.; Speicher, D.W. Electrophoretic prefractionation for comprehensive analysis of proteomes. In Proteome Analysis: Interpreting the Genome; Elsevier: Amsterdam, The Netherlands, 2004; pp. 93–118. [Google Scholar]
- Hoving, S.; Voshol, H.; van Oostrum, J. Towards high performance two-dimensional gel electrophoresis using ultrazoom gels. Electrophoresis 2000, 21, 2617–2621. [Google Scholar] [CrossRef]
- Richardson, M.R.; Liu, S.; Ringham, H.N.; Chan, V.; Witzmann, F.A. Sample complexity reduction for two-dimensional electrophoresis using solution isoelectric focusing prefractionation. Electrophoresis 2008, 29, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Oguri, T.; Takahata, I.; Katsuta, K.; Nomura, E.; Hidaka, M.; Inagaki, N. Proteome analysis of rat hippocampal neurons by multiple large gel two-dimensional electrophoresis. Proteomics 2002, 2, 666–672. [Google Scholar] [CrossRef]
- Westermeier, R.; Naven, T. Proteomics in Practice: A Laboratory Manual of Proteome Analysis; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002. [Google Scholar]
- Miller, I.; Crawford, J.; Gianazza, E. Protein stains for proteomic applications: Which, when, why? Proteomics 2006, 6, 5385–5408. [Google Scholar] [CrossRef] [PubMed]
- Gauci, V.J.; Wright, E.P.; Coorssen, J.R. Quantitative proteomics: Assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 2011, 4, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, T.H. Protein gel staining methods: An introduction and overview. Methods Enzymol. 2009, 463, 541–563. [Google Scholar] [PubMed]
- Patton, W.F. Detection technologies in proteome analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 771, 3–31. [Google Scholar] [CrossRef]
- Rabilloud, T.; Vuillard, L.; Gilly, C.; Lawrence, J.J. Silver-staining of proteins in polyacrylamide gels: A general overview. Cell. Mol. Biol. 1994, 40, 57–75. [Google Scholar] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, F.; Centeno, D.; Rofidal, V.; Tauzin, M.; Martin, O.; Sonamerer, N.; Rossignol, M. Different impact of staining procedures using visible stains and fluorescent dyes for large-scale investigation of proteomes by MALDI-TOF mass spectrometry. J. Proteome Res. 2006, 5, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Patton, W.F. A thousand points of light: The application of fluorescence detection technologies to two-dimensional gel electrophoresis and proteomics. Electrophoresis 2000, 21, 1123–1144. [Google Scholar] [CrossRef]
- Unlü, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteom. 2011, 74, 1829–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabilloud, T. Two-dimensional gel electrophoresis in proteomics: Old, old fashioned, but it still climbs up the mountains. Proteomics 2002, 2, 3–10. [Google Scholar] [CrossRef]
- Braun, R.J.; Kinkl, N.; Beer, M.; Ueffing, M. Two-dimensional electrophoresis of membrane proteins. Anal. Bioanal. Chem. 2007, 389, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Celis, J.E. A search for differential polypeptide synthesis throughout the cell cycle of HeLa cells. J. Cell Biol. 1980, 84, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Fey, S.J.; Bellatin, J.; Larsen, P.M.; Arevalo, J.; Celis, J.E. Identification of a nuclear and of a cytoplasmic polypeptide whose relative proportions are sensitive to changes in the rate of cell proliferation. Exp. Cell Res. 1981, 136, 311–319. [Google Scholar] [CrossRef]
- Bravo, R.; Frank, R.; Blundell, P.A.; Macdonald-Bravo, H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta. Nature 1987, 326, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N. Proliferating cell nuclear antigen: A proteomics view. Cell. Mol. Life Sci. 2008, 65, 3789–3808. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Lee, H. Characterization of proliferating cell nuclear antigen (PCNA) isoforms in normal and cancer cells: There is no cancer-associated form of PCNA. FEBS Lett. 2007, 581, 4917–4920. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Lee, H. The post-translational modifications of proliferating cell nuclear antigen: Acetylation, not phosphorylation, plays an important role in the regulation of its function. J. Biol. Chem. 2004, 279, 20194–20199. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Lee, H. Observation of multiple isoforms and specific proteolysis patterns of proliferating cell nuclear antigen in the context of cell cycle compartments and sample preparations. Proteomics 2003, 3, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Q.; Chen, X.-Z. Detecting protein-protein interactions by Far western blotting. Nat. Protoc. 2007, 2, 3278–3284. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Lee, H. Proliferating cell nuclear antigen in the cytoplasm interacts with components of glycolysis and cancer. FEBS Lett. 2010, 584, 4292–4298. [Google Scholar] [CrossRef] [PubMed]
- Guichet, A.; Copeland, J.W.; Erdélyi, M.; Hlousek, D.; Závorszky, P.; Ho, J.; Brown, S.; Percival-Smith, A.; Krause, H.M.; Ephrussi, A. The nuclear receptor homologue Ftz-F1 and the homeodomain protein Ftz are mutually dependent cofactors. Nature 1997, 385, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Webster, J.; Oxley, D. Protein Identification by MALDI-TOF mass spectrometry. Methods Mol. Biol. 2012, 800, 227–240. [Google Scholar] [PubMed]
- Pappin, D.J.C.; Hojrup, P.; Bleasby, A.J. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993, 3, 327–332. [Google Scholar] [CrossRef]
- Henzel, W.J.; Billeci, T.M.; Stults, J.T.; Wong, S.C.; Grimley, C.; Watanabe, C. Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc. Natl. Acad. Sci. USA 1993, 90, 5011–5015. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Hojrup, P.; Roepstorff, P. Use of mass spectrometric molecular weight information to identify proteins in sequence databases. Biol. Mass Spectrom. 1993, 22, 338–345. [Google Scholar] [CrossRef] [PubMed]
- James, P.; Quadroni, M.; Carafoli, E.; Gonnet, G. Protein identification by mass profile fingerprinting. Biochem. Biophys. Res. Commun. 1993, 195, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Speicher, S.; Griffin, P.R.; Hunkapiller, T. Peptide Mass Maps: A Highly Informative Approach to Protein Identification. Anal. Biochem. 1993, 214, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ueberle, B.; Frank, R.; Herrmann, R. The proteome of the bacterium Mycoplasma pneumoniae: Comparing predicted open reading frames to identified gene products. Proteomics 2002, 2, 754–764. [Google Scholar] [CrossRef]
- Jungblut, P.R.; Hecker, M. Proteomics of Microbial Pathogens; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2007. [Google Scholar]
- Naryzhny, S.N.; Lisitsa, A.V.; Zgoda, V.G.; Ponomarenko, E.A.; Archakov, A.I. 2DE-based approach for estimation of number of protein species in a cell. Electrophoresis 2014, 35, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Zgoda, V.G.; Maynskova, M.A.; Ronzhina, N.L.; Belyakova, N.V.; Legina, O.K.; Archakov, A.I. Experimental estimation of proteome size for cells and human plasma. Biomed. Khim. 2015, 61, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. Protein species high resolution quantitative proteomics of HeLa cells using SILAC-2-DE-nanoLC/LTQ-Orbitrap mass spectrometry. Mol. Cell. Proteom. 2012, 12, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Zgoda, V.G.; Maynskova, M.A.; Novikova, S.E.; Ronzhina, N.L.; Vakhrushev, I.V.; Khryapova, E.V.; Lisitsa, A.V.; Tikhonova, O.V.; Ponomarenko, E.A.; et al. Combination of virtual and experimental 2DE together with ESI LC-MS/MS gives a clearer view about proteomes of human cells and plasma. Electrophoresis 2016, 37, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Maynskova, M.A.; Zgoda, V.G.; Ronzhina, N.L.; Kleyst, O.A.; Vakhrushev, I.V.; Archakov, A.I. Virtual-Experimental 2DE Approach in Chromosome-Centric Human Proteome Project. J. Proteome Res. 2016, 15, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Maynskova, M.A.; Zgoda, V.G.; Ronzhina, N.L.; Novikova, S.E. Proteomic profiling of high-grade glioblastoma using virtual-experimental 2DE. J. Proteom. Bioinform. 2016, 9, 158–165. [Google Scholar] [CrossRef]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially Modified Protein Abundance Index (emPAI) for Estimation of Absolute Protein Amount in Proteomics by the Number of Sequenced Peptides per Protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Kusebauch, U.; Campbell, D.S.; Deutsch, E.W.; Chu, C.S.; Spicer, D.A.; Brusniak, M.Y.; Slagel, J.; Sun, Z.; Stevens, J.; Grimes, B.; et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell 2016, 166, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Blein-Nicolas, M.; Zivy, M. Thousand and one ways to quantify and compare protein abundances in label-free bottom-up proteomics. Biochim. Biophys. Acta Proteins Proteom. 2015, 1864, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Osborn, M. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 1969, 244, 4406–4412. [Google Scholar] [PubMed]
- Appel, R.D.; Sanchez, J.C.; Bairoch, A.; Golaz, O.; Miu, M.; Vargas, J.R.; Hochstrasser, D.F. SWISS-2DPAGE: A database of two-dimensional gel electrophoresis images. Electrophoresis 1993, 14, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Appel, R.D.; Sanchez, J.C.; Bairoch, A.; Golaz, O.; Ravier, F.; Pasquali, C.; Hughes, G.J.; Hochstrasser, D.F. The SWISS-2DPAGE database of two-dimensional polyacrylamide gel electrophoresis. Nucleic Acids Res. 1994, 22, 3581–3582. [Google Scholar] [PubMed]
- Appel, R.D.; Bairoch, A.; Sanchez, J.C.; Vargas, J.R.; Golaz, O.; Pasquali, C.; Hochstrasser, D.F. Federated two-dimensional electrophoresis database: A simple means of publishing two-dimensional electrophoresis data. Electrophoresis 1996, 17, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Giometti, C.S.; Williams, K.; Tollaksen, S.L. A two-dimensional electrophoresis database of human breast epithelial cell proteins. Electrophoresis 1997, 18, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, D.F.; Frutiger, S.; Paquet, N.; Bairoch, A.; Ravier, F.; Pasquali, C.; Sanchez, J.C.; Tissot, J.D.; Bjellqvist, B.; Vargas, R. Human liver protein map: A reference database established by microsequencing and gel comparison. Electrophoresis 1992, 13, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.J.; Frutiger, S.; Paquet, N.; Pasquali, C.; Sanchez, J.C.; Tissot, J.D.; Bairoch, A.; Appel, R.D.; Hochstrasser, D.F. Human liver protein map: Update 1993. Electrophoresis 1993, 14, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Golaz, O.; Hughes, G.J.; Frutiger, S.; Paquet, N.; Bairoch, A.; Pasquali, C.; Sanchez, J.C.; Tissot, J.D.; Appel, R.D.; Walzer, C. Plasma and red blood cell protein maps: Update 1993. Electrophoresis 1993, 14, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Wu, W.; Hood, L.; Harrington, M. Human cerebrospinal fluid protein database: Edition 1992. Electrophoresis 1992, 13, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- VanBogelen, R.A.; Sankar, P.; Clark, R.L.; Bogan, J.A.; Neidhardt, F.C. The gene-protein database of Escherichia coli: Edition 5. Electrophoresis 1992, 13, 1014–1054. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, C.; Frutiger, S.; Wilkins, M.R.; Hughes, G.J.; Appel, R.D.; Bairoch, A.; Schaller, D.; Sanchez, J.C.; Hochstrasser, D.F. Two-dimensional gel electrophoresis of Escherichia coli homogenates: The Escherichia coli SWISS-2DPAGE database. Electrophoresis 1996, 17, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Garrels, J.I.; Franza, B.R. The REF52 protein database. Methods of database construction and analysis using the QUEST system and characterizations of protein patterns from proliferating and quiescent REF52 cells. J. Biol. Chem. 1989, 264, 5283–5298. [Google Scholar] [PubMed]
- Latham, K.E.; Garrels, J.I.; Chang, C.; Solter, D. Analysis of embryonic mouse development: Construction of a high-resolution, two-dimensional gel protein database. Appl. Theor. Electrophor. 1992, 2, 163–170. [Google Scholar] [PubMed]
- Rasmussen, H.H.; van Damme, J.; Puype, M.; Gesser, B.; Celis, J.E.; Vandekerckhove, J. Microsequences of 145 proteins recorded in the two-dimensional gel protein database of normal human epidermal keratinocytes. Electrophoresis 1992, 13, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Giometti, C.S.; Taylor, J.; Tollaksen, S.L. Mouse liver protein database: A catalog of proteins detected by two-dimensional gel electrophoresis. Electrophoresis 1992, 13, 970–991. [Google Scholar] [CrossRef] [PubMed]
- Bjellqvist, B.; Hughes, G.J.; Pasquali, C.; Paquet, N.; Ravier, F.; Sanchez, J.C.; Frutiger, S.; Hochstrasser, D. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis 1993, 14, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Schobert, M.; Hundertmark, C.; Jahn, D.; Münch, R. JVirGel: Calculation of virtual two-dimensional protein gels. Nucleic Acids Res. 2003, 31, 3862–3865. [Google Scholar] [CrossRef] [PubMed]
- Medjahed, D.; Smythers, G.W.; Powell, D.A.; Stephens, R.M.; Lemkin, P.F.; Munroe, D.J. VIRTUAL2D: A web-accessible predictive, database for proteomics analysis. Proteomics 2003, 3, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Virtual Gel. Available online: http://www.poirrier.be/~jean-etienne/software/vgel/ (accessed on 12 November 2016).
- JVirGel. Available online: http://www.jvirgel.de/ (accessed on 12 November 2016).
- Medjahed, D.; Luke, B.T.; Tontesh, T.S.; Smythers, G.W.; Munroe, D.J.; Lemkin, P.F. Tissue molecular anatomy project (TMAP): An expression database for comparative cancer proteomics. Proteomics 2003, 3, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Medjahed, D.; Lemkin, P.F.; Smythers, G.W.; Munroe, D.J. Looking for cancer clues in publicly accessible databases. Comp. Funct. Genom. 2004, 5, 196–200. [Google Scholar] [CrossRef] [PubMed]
- ProMoST: Protein Modification Screening Tool. Available online: http://proteomics.mcw.edu/promost.html (accessed on 12 November 2016).
- Halligan, B.D. ProMoST: A tool for calculating the pI and molecular mass of phosphorylated and modified proteins on two-dimensional gels. Methods Mol. Biol. 2009, 527, 283–298. [Google Scholar] [PubMed]
- Ogorzalek Loo, R.R.; Stevenson, T.I.; Mitchell, C.; Loo, J.A.; Andrews, P.C. Mass spectrometry of proteins directly from polyacrylamide gels. Anal. Chem. 1996, 68, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Loo, R.R.; Cavalcoli, J.D.; VanBogelen, R.A.; Mitchell, C.; Loo, J.A; Moldover, B.; Andrews, P.C. Virtual 2-D gel electrophoresis: Visualization and analysis of the E. coli proteome by mass spectrometry. Anal. Chem. 2001, 73, 4063–4070. [Google Scholar] [PubMed]
- Lohnes, K.; Quebbemann, N.R.; Liu, K.; Kobzeff, F.; Loo, J.A.; Ogorzalek Loo, R.R. Combining high-throughput MALDI-TOF mass spectrometry and isoelectric focusing gel electrophoresis for virtual 2D gel-based proteomics. Methods 2016, 104, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, C.; Mostaguir, K.; Sanchez, J.C.; Hochstrasser, D.F.; Appel, R.D. SWISS-2DPAGE, ten years later. Proteomics 2004, 4, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Compute pI/Mw Tool. Available online: http://web.expasy.org/compute_pi/pi_tool-doc.html (accessed on 12 November 2016).
- Archakov, A.; Aseev, A.; Bykov, V.; Grigoriev, A.; Govorun, V.; Ivanov, V.; Khlunov, A.; Lisitsa, A.; Mazurenko, S.; Makarov, A.A.; et al. Gene-centric view on the human proteome project: The example of the Russian roadmap for chromosome 18. Proteomics 2011, 11, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Ziemer, M.A.; Mason, A.; Carlson, D.M. Cell-free translations of proline-rich protein mRNAs. J. Biol. Chem. 1982, 257, 11176–11180. [Google Scholar] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naryzhny, S. Towards the Full Realization of 2DE Power. Proteomes 2016, 4, 33. https://doi.org/10.3390/proteomes4040033
Naryzhny S. Towards the Full Realization of 2DE Power. Proteomes. 2016; 4(4):33. https://doi.org/10.3390/proteomes4040033
Chicago/Turabian StyleNaryzhny, Stanislav. 2016. "Towards the Full Realization of 2DE Power" Proteomes 4, no. 4: 33. https://doi.org/10.3390/proteomes4040033
APA StyleNaryzhny, S. (2016). Towards the Full Realization of 2DE Power. Proteomes, 4(4), 33. https://doi.org/10.3390/proteomes4040033