
A Quantitative Proteomics Approach to Clinical Research with Non-Traditional Samples


Abstract
:1. Introduction
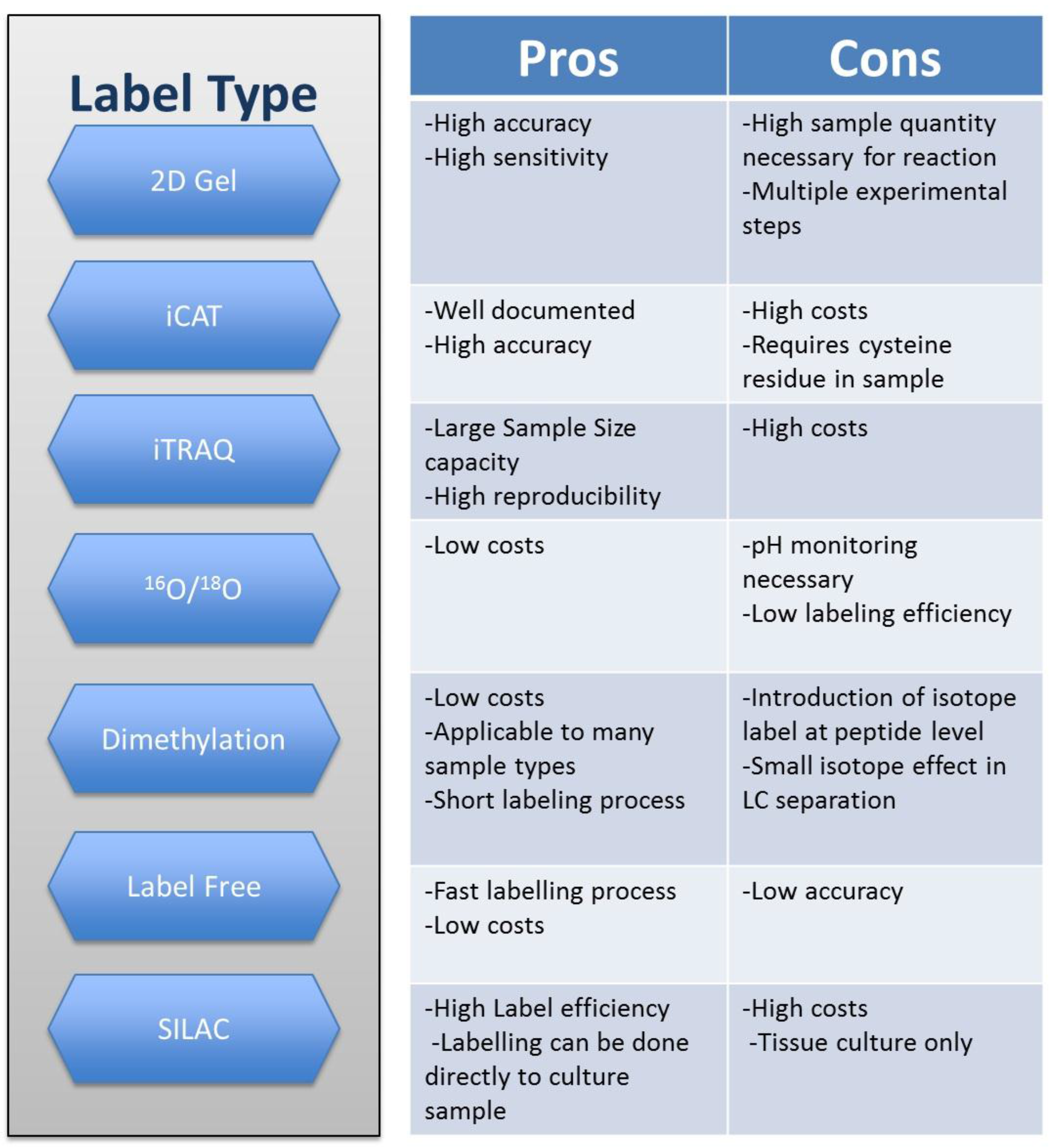
2. Techniques in Quantitative Proteomics
3. Ear Wax “Cerumen”
4. Saliva
5. Vitreous Humor
6. Aqueous Humor
7. Tears
8. Nipple Aspirate Fluid
9. Breast Milk/Colostrum
10. Cervicovaginal Fluid
11. Nasal Secretions
12. Broncho Alveolar Lavage Fluid
13. Stools
14. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Semba, R.D.; Enghild, J.J.; Venkatraman, V.; Dyrlund, T.F.; Eyk, J.E. The human eye proteome project: Perspectives on an emerging proteome. Proteomics 2013, 13, 2500–2511. [Google Scholar] [CrossRef] [PubMed]
- Bennike, T.; Birkelund, S.; Stensballe, A.; Andersen, V. Biomarkers in inflammatory bowel diseases: Current status and proteomics identification strategies. World J. Gastroenterol 2014, 20, 3231–3244. [Google Scholar] [CrossRef] [PubMed]
- Feig, M.A.; Hammer, E.; Volker, U.; Jehmlich, N. In-depth proteomic analysis of the human cerumen-a potential novel diagnostically relevant biofluid. J. Proteom. 2013, 83, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Vestling, M.M. Using mass spectrometry for proteins. J. Chem. Educ. 2003, 80, 122. [Google Scholar] [CrossRef]
- Bonzon-Kulichenko, E.; Pérez-Hernández, D.; Núñez, E.; Martínez-Acedo, P.; Navarro, P.; Trevisan-Herraz, M.; del Carmen Ramos, M.; Sierra, S.; Martínez-Martínez, S.; Ruiz-Meana, M. A robust method for quantitative high-throughput analysis of proteomes by 18O labeling. Mol. Cell. Proteom. 2011, 10, M110.003335. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Biomarkers in risk assessment: Validity and validation. In Environmental Health Criteria; WHO: Geneva, Switzerland, 2001; Volume 222. [Google Scholar]
- Chahrour, O.; Cobice, D.; Malone, J. Stable isotope labelling methods in mass spectrometry-based quantitative proteomics. J. Pharm. Biomed. Anal. 2015, 113, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Lam, C.; Chan, M.; Cheung, R.; Law, L.; Lit, L.; Ng, K.; Suen, M.; Tai, H. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clinic. Biochem. Rev. 2003, 24, 3–12. [Google Scholar]
- Andersson, M.; Andren, P.; Caprioli, R. Maldi imaging and profiling mass spectrometry in neuroproteomics. In Neuroproteomics; Alzate, O., Ed.; CRC Press/Taylor and Francis: Boca Raton, FL, USA, 2010; Chapter 7. [Google Scholar]
- Megger, D.A.; Bracht, T.; Meyer, H.E.; Sitek, B. Label-free quantification in clinical proteomics. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2013, 1834, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Kito, K.; Ito, T. Mass spectrometry-based approaches toward absolute quantitative proteomics. Curr. Genom. 2008, 9, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Boersema, P.J.; Raijmakers, R.; Lemeer, S.; Mohammed, S.; Heck, A.J. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009, 4, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yu, T.; Xie, Y.; Yang, Y.; Li, Y.; Zhou, X.; Tsung, S.; Loo, R.R.; Loo, J.A.; Wong, D.T. Discovery of oral fluid biomarkers for human oral cancer by mass spectrometry. Cancer Genom.-Proteom. 2007, 4, 55–64. [Google Scholar]
- De Jong, E.P.; Xie, H.; Onsongo, G.; Stone, M.D.; Chen, X.B.; Kooren, J.A.; Refsland, E.W.; Griffin, R.J.; Ondrey, F.G.; Wu, B. Quantitative proteomics reveals myosin and actin as promising saliva biomarkers for distinguishing pre-malignant and malignant oral lesions. PLoS ONE 2010, 5, e11148. [Google Scholar] [CrossRef] [PubMed]
- Bassim, C.W.; Ambatipudi, K.S.; Mays, J.W.; Edwards, D.A.; Swatkoski, S.; Fassil, H.; Baird, K.; Gucek, M.; Melvin, J.E.; Pavletic, S.Z. Quantitative salivary proteomic differences in oral chronic graft-versus-host disease. J. Clin. Immunol. 2012, 32, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Devic, I.; Shi, M.; Schubert, M.M.; Lloid, M.; Izutsu, K.T.; Pan, C.; Missaghi, M.; Morton, T.H.; Mancl, L.A.; Zhang, J. Proteomic analysis of saliva from patients with oral chronic graft-versus-host disease. Biol. Blood Marrow Transplant. 2014, 20, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Ambatipudi, K.S.; Swatkoski, S.; Moresco, J.J.; Tu, P.G.; Coca, A.; Anolik, J.H.; Gucek, M.; Sanz, I.; Yates, J.R.; Melvin, J.E. Quantitative proteomics of parotid saliva in primary sjögren’s syndrome. Proteomics 2012, 12, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, J.; Meijer, J.; Ieong, S.; Xie, Y.; Yu, T.; Zhou, H.; Henry, S.; Vissink, A.; Pijpe, J. Salivary proteomic and genomic biomarkers for primary sjögren’s syndrome. Arthr. Rheum. 2007, 56, 3588–3600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Z.; Feng, S.; Wang, Q.; Malamud, D.; Deng, H. Quantitative analysis of differentially expressed saliva proteins in human immunodeficiency virus type 1 (HIV-1) infected individuals. Anal. Chim. Acta 2013, 774, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Brown, J.N.; Ryder, M.I.; Gritsenko, M.; Jacobs, J.M.; Smith, R.D. Proteomic analysis of saliva in hiv-positive heroin addicts reveals proteins correlated with cognition. PLoS ONE 2014, 9, e89366. [Google Scholar] [CrossRef] [PubMed]
- Bencharit, S.; Baxter, S.S.; Carlson, J.; Byrd, W.C.; Mayo, M.V.; Border, M.B.; Kohltfarber, H.; Urrutia, E.; Howard-Williams, E.L.; Offenbacher, S. Salivary proteins associated with hyperglycemia in diabetes: A proteomic analysis. Mol. BioSyst. 2013, 9, 2785–2797. [Google Scholar] [CrossRef] [PubMed]
- Cabras, T.; Pisano, E.; Montaldo, C.; Giuca, M.R.; Iavarone, F.; Zampino, G.; Castagnola, M.; Messana, I. Significant modifications of the salivary proteome potentially associated with complications of down syndrome revealed by top-down proteomics. Mol. Cell. Proteom. 2013, 12, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Krohne, T.U.; Kammerer, K.; Warnken, U.; Hotz-Wagenblatt, A.; Bergmann, M.; Stanzel, B.V.; Kempf, T.; Holz, F.G.; Schnölzer, M. In-depth mass spectrometric mapping of the human vitreous proteome. Proteom. Sci. 2013, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Skeie, J.M.; Roybal, C.N.; Mahajan, V.B. Proteomic insight into the molecular function of the vitreous. PLoS ONE 2015, 10, e0127567. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.-B.; Chen, X.; Timothy, N.; Aiello, L.P.; Feener, E.P. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J. Proteom. Res. 2008, 7, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, S.J.; Yu, H.G.; Yu, J.; Park, K.S.; Jang, I.J.; Kim, Y. Verification of biomarkers for diabetic retinopathy by multiple reaction monitoring. J. Proteom. Res. 2009, 9, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, L.; Hu, J.W.; Xie, C.L.; Wang, F. Characterisation of the vitreous proteome in proliferative diabetic retinopathy. Proteom. Sci. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, L.; Hu, J.; Xie, C.; Wang, F. Differentiating vitreous proteomes in proliferative diabetic retinopathy using high-performance liquid chromatography coupled to tandem mass spectrometry. Exp. Eye Res. 2013, 108, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Loukovaara, S.; Nurkkala, H.; Tamene, F.; Gucciardo, E.; Liu, X.; Repo, P.; Lehti, K.; Varjosalo, M. Quantitative proteomics analysis of vitreous humor from diabetic retinopathy patients. J. Proteom. Res. 2015, 14, 5131–5143. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Min, H.; Kim, S.J.; Oh, S.; Kim, K.; Yu, H.G.; Park, T.; Kim, Y. Development of diagnostic biomarkers for detecting diabetic retinopathy at early stages using quantitative proteomics. J. Diabetes Rev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Koss, M.J.; Hoffmann, J.; Nguyen, N.; Pfister, M.; Mischak, H.; Mullen, W.; Husi, H.; Rejdak, R.; Koch, F.; Jankowski, J. Proteomics of vitreous humor of patients with exudative age-related macular degeneration. PLoS ONE 2014, 9, e96895. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Feng, L.; Wu, Y.; Wang, H.; Ba, J.; Zhu, W.; Xie, C. Vitreous proteomic analysis of idiopathic epiretinal membranes. Mol. BioSyst. 2014, 10, 2558–2566. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kang, J.W.; Ahn, J.; Lee, E.K.; Cho, K.-C.; Han, B.N.R.; Hong, N.Y.; Park, J.; Kim, K.P. Proteomic analysis of the aqueous humor in age-related macular degeneration (amd) patients. J. Proteom. Res. 2012, 11, 4034–4043. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Choi, A.J.; Kang, G.-Y.; Park, H.S.; Kim, H.C.; Lim, H.J.; Chung, H. Increased 26s proteasome non-atpase regulatory subunit 1 in the aqueous humor of patients with age-related macular degeneration. BMB Rep. 2014, 47, 292. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Liu, X.; Yang, Q.; Zhuang, M.; Wang, F.; Chen, X.; Hang, H.; Zhang, W.; Liu, Q. Proteomic analysis of the aqueous humor in patients with wet age-related macular degeneration. Proroteom.-Chin Appl. 2013, 7, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.; Villarrubia, A.; Merayo-Lloves, J.; Elortza, F.; Azkargorta, M.; de Toledo, J.A.; Rodriguez-Agirretxe, I.; Suarez, T.; Acera, A. Label-free LC–MS/MS quantitative analysis of aqueous humor from keratoconic and normal eyes. Mol. Vis. 2015, 21, 451. [Google Scholar] [PubMed]
- Ayuso, V.K.; de Boer, J.H.; Byers, H.L.; Coulton, G.R.; Dekkers, J.; de Visser, L.; van Loon, A.M.; Schellekens, P.A.; Rothova, A.; de Groot-Mijnes, J.D. Intraocular biomarker identification in uveitis associated with juvenile idiopathic arthritisjia-associated uveitis biomarker identification. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3709–3720. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.-Y.; Tsai, M.-L.; Wang, C.-Y.; Chen, A.; Chou, Y.-C.; Hsia, C.-W.; Wu, Y.-F.; Chen, H.-M.; Huang, T.-H.; Chen, P.-H. Proteomic analysis and identification of aqueous humor proteins with a pathophysiological role in diabetic retinopathy. J. Proteom. 2012, 75, 2950–2959. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Chen, Z.; Yang, Q.; Liu, X.; Chen, X.; Zhuang, M.; Liu, Q. Proteomic analysis of aqueous humor from patients with branch retinal vein occlusion-induced macular edema. Int. J. Mol. Med. 2013, 32, 1421–1434. [Google Scholar] [PubMed]
- Zhou, L.; Beuerman, R.W.; Chan, C.M.; Zhao, S.Z.; Li, X.R.; Yang, H.; Tong, L.; Liu, S.; Stern, M.E.; Tan, D. Identification of tear fluid biomarkers in dry eye syndrome using itraq quantitative proteomics. J. Proteom. Res. 2009, 8, 4889–4905. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Thangavelu, M.; Zhang, L.; Green, K.B.; Nichols, K.K. Itraq quantitative proteomics in the analysis of tears in dry eye patientsanalysis of tears in dry eye patients. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5052–5059. [Google Scholar] [CrossRef] [PubMed]
- Boehm, N.; Funke, S.; Wiegand, M.; Wehrwein, N.; Pfeiffer, N.; Grus, F.H. Alterations in the tear proteome of dry eye patients—a matter of the clinical phenotypetear proteome of dry eye patients. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.J.; Green-Church, K.B. Mass spectrometry-based proteomic analyses in contact lens-related dry eye. Cornea 2009, 28, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sheng, M.; Xie, L.; Liu, F.; Yan, G.; Wang, W.; Lin, A.; Zhao, F.; Chen, Y. Tear proteomic analysis of patients with type 2 diabetes and dry eye syndrome by two-dimensional nano-liquid chromatography coupled with tandem mass spectrometrynano-liquid chromatography/tandem mass spectrometry. Investig. Ophthalmol. Vis. Sci. 2014, 55, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Csősz, É.; Boross, P.; Csutak, A.; Berta, A.; Tóth, F.; Póliska, S.; Török, Z.; Tőzsér, J. Quantitative analysis of proteins in the tear fluid of patients with diabetic retinopathy. J. Proteom. 2012, 75, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Matheis, N.; Grus, F.H.; Breitenfeld, M.; Knych, I.; Funke, S.; Pitz, S.; Ponto, K.A.; Pfeiffer, N.; Kahaly, G.J. Proteomics differentiate between thyroid-associated orbitopathy and dry eye syndromeproteomics of tears. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; Bucci, S.; Agnifili, L.; Fasanella, V.; D'Aguanno, S.; Mastropasqua, A.; Ciancaglini, M.; Mastropasqua, L.; di Ilio, C.; Sacchetta, P. Differential protein expression in tears of patients with primary open angle and pseudoexfoliative glaucoma. Mol. BioSyst. 2012, 8, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Kalló, G.; Emri, M.; Varga, Z.; Ujhelyi, B.; Tőzsér, J.; Csutak, A.; Csősz, É. Changes in the chemical barrier composition of tears in alzheimer’s disease reveal potential tear diagnostic biomarkers. PLoS ONE 2016, 11, e0158000. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Petznick, A.; Tong, L.; Hall, R.C.; Rosman, M.; Chan, C.; Koh, S.K.; Beuerman, R.W.; Zhou, L.; Mehta, J.S. Comparative analysis of two femtosecond lasik platforms using itraq quantitative proteomicstear protein profile in lasik. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3396–3402. [Google Scholar] [CrossRef] [PubMed]
- Salvisberg, C.; Tajouri, N.; Hainard, A.; Burkhard, P.R.; Lalive, P.H.; Turck, N. Exploring the human tear fluid: Discovery of new biomarkers in multiple sclerosis. Proteom.-Chin Appl. 2014, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Pieragostino, D.; Agnifili, L.; Fasanella, V.; D'Aguanno, S.; Mastropasqua, R.; Di Ilio, C.; Sacchetta, P.; Urbani, A.; Del Boccio, P. Shotgun proteomics reveals specific modulated protein patterns in tears of patients with primary open angle glaucoma naive to therapy. Mol. BioSyst. 2013, 9, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Zangar, R.C.; Varnum, S.M.; Covington, C.Y.; Smith, R.D. A rational approach for discovering and validating cancer markers in very small samples using mass spectrometry and elisa microarrays. Dis. Markers 2004, 20, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Brunoro, G.V.F.; Carvalho, P.C.; da Silva Ferreira, A.T.; Perales, J.; Valente, R.H.; de Moura Gallo, C.V.; Pagnoncelli, D.; da Costa Neves-Ferreira, A.G. Proteomic profiling of nipple aspirate fluid (NAF): Exploring the complementarity of different peptide fractionation strategies. J. Proteom. 2015, 117, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Paweletz, C.P.; Trock, B.; Pennanen, M.; Tsangaris, T.; Magnant, C.; Liotta, L.A.; Petricoin III, E.F. Proteomic patterns of nipple aspirate fluids obtained by seldi-tof: Potential for new biomarkers to aid in the diagnosis of breast cancer. Dis. Markers 2001, 17, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Alexander, H.; Stegner, A.L.; Wagner-Mann, C.; du Bois, G.C.; Alexander, S.; Sauter, E.R. Proteomic analysis to identify breast cancer biomarkers in nipple aspirate fluid. Chin. Cancer Res. 2004, 10, 7500–7510. [Google Scholar] [CrossRef] [PubMed]
- Sauter, E.R.; Shan, S.; Hewett, J.E.; Speckman, P.; du Bois, G.C. Proteomic analysis of nipple aspirate fluid using seldi-tof-ms. Int. J. Cancer 2005, 114, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Loud, J.T.; Gierach, G.L.; Veenstra, T.D.; Falk, R.T.; Nichols, K.; Guttmann, A.; Xu, X.; Greene, M.H.; Gail, M.H. Circulating estrogens and estrogens within the breast among postmenopausal BRCA1/2 mutation carriers. Breast Cancer Res. Treat. 2014, 143, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Coscia, A.; Orrù, S.; Di Nicola, P.; Giuliani, F.; Rovelli, I.; Peila, C.; Martano, C.; Chiale, F.; Bertino, E. Cow’s milk proteins in human milk. J. Biol. Regul. Homeost. Agents 2012, 26, 39–42. [Google Scholar] [PubMed]
- Coscia, A.; Orrù, S.; Di Nicola, P.; Giuliani, F.; Varalda, A.; Peila, C.; Fabris, C.; Conti, A.; Bertino, E. Detection of cow’s milk proteins and minor components in human milk using proteomics techniques. J. Matern.-Fetal Neonatal Med. 2012, 25, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Alvarado, R.; Phinney, B.; Lonnerdal, B. Proteomic characterization of human milk whey proteins during a twelve-month lactation period. J. Proteom. Res. 2011, 10, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Alvarado, R.; Phinney, B.; Lonnerdal, B. Proteomic characterization of specific minor proteins in the human milk casein fraction. J. Proteom. Res. 2011, 10, 5409–5415. [Google Scholar] [CrossRef] [PubMed]
- Grapov, D.; Lemay, D.G.; Weber, D.; Phinney, B.S.; Azulay Chertok, I.R.; Gho, D.S.; German, J.B.; Smilowitz, J.T. The human colostrum whey proteome is altered in gestational diabetes mellitus. J. Proteom. Res. 2014, 14, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.L.; Smith, C.R.; Diamandis, E.P. Proteomic analysis of human cervico-vaginal fluid. J. Proteom. Res. 2007, 6, 2859–2865. [Google Scholar] [CrossRef] [PubMed]
- Di Quinzio, M.K.; Oliva, K.; Holdsworth, S.J.; Ayhan, M.; Walker, S.P.; Rice, G.E.; Georgiou, H.M.; Permezel, M. Proteomic analysis and characterisation of human cervico-vaginal fluid proteins. Aust. N. Z. J. Obstet. Gynaecol. 2007, 47, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, N.; Cole, A.L.; Svoboda, P.; Pohl, J.; Cole, A.M. Cationic polypeptides are required for anti-hiv-1 activity of human vaginal fluid. J. Immunol. 2005, 175, 7560–7567. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, G.; Zegels, G.; Coen, E.; Vuylsteke, B.; Jennes, W.; Van Ostade, X. Increased serpin A5 levels in the cervicovaginal fluid of HIV-1 exposed seronegatives suggest that a subtle balance between serine proteases and their inhibitors may determine susceptibility to HIV-1 infection. Virology 2014, 458, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, G.A.; Tjalma, W.A.; Coen, E.P.; Depuydt, C.E.; Van Ostade, X.W. Identification of protein biomarkers for cervical cancer using human cervicovaginal fluid. PLoS ONE 2014, 9, e106488. [Google Scholar] [CrossRef] [PubMed]
- Debat, H.; Eloit, C.; Blon, F.; Sarazin, B.; Henry, C.; Huet, J.-C.; Trotier, D.; Pernollet, J.-C. Identification of human olfactory cleft mucus proteins using proteomic analysis. J. Proteom. Res. 2007, 6, 1985–1996. [Google Scholar] [CrossRef] [PubMed]
- Casado, B.; Pannell, L.K.; Iadarola, P.; Baraniuk, J.N. Identification of human nasal mucous proteins using proteomics. Proteomics 2005, 5, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Saieg, A.; Brown, K.J.; Pena, M.T.; Rose, M.C.; Preciado, D. Proteomic analysis of pediatric sinonasal secretions shows increased muc5b mucin in crs. Pediatr. Res. 2014, 77, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Sakamoto, N.; Ishimatsu, Y.; Kakugawa, T.; Nakashima, S.; Hara, S.; Adachi, M.; Fujita, H.; Mukae, H.; Kohno, S. S100a9 in balf is a candidate biomarker of idiopathic pulmonary fibrosis. Respir. Med. 2012, 106, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; Morrison, L.D.; Todd, J.L.; Snyder, L.D.; Thompson, J.W.; Soderblom, E.J.; Plonk, K.; Weinhold, K.J.; Townsend, R.; Minnich, A. Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J. Proteom. Res. 2015, 14, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Mammen, M.J.; Li, J.; Shen, X.; Jiang, X.; Hu, Q.; Wang, J.; Sethi, S.; Qu, J. Large-scale, ion-current-based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J. Proteom. Res. 2013, 13, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Pastor, M.; Nogal, A.; Molina-Pinelo, S.; Melendez, R.; Salinas, A.; De la Pena, M.G.; Martin-Juan, J.; Corral, J.; Garcia-Carbonero, R.; Carnero, A. Identification of proteomic signatures associated with lung cancer and copd. J. Proteom. 2013, 89, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, M.; Becker, T.L.; Viken, K.J.; Jagtap, P.D.; Dey, S.; Steinbach, M.S.; Wu, B.; Kumar, V.; Bitterman, P.B.; Ingbar, D.H. Proteomic profiles in acute respiratory distress syndrome differentiates survivors from non-survivors. PLoS ONE 2014, 9, e109713. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; McDonald, C.F.; Collins, A.L.; Darby, I.A.; Pouniotis, D.S. Quantitative proteomics of bronchoalveolar lavage fluid in lung adenocarcinoma. Cancer Genom.-Proteom. 2015, 12, 39–48. [Google Scholar]
- Verberkmoes, N.C.; Russell, A.L.; Shah, M.; Godzik, A.; Rosenquist, M.; Halfvarson, J.; Lefsrud, M.G.; Apajalahti, J.; Tysk, C.; Hettich, R.L. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009, 3, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Kolmeder, C.A.; De Been, M.; Nikkilä, J.; Ritamo, I.; Mättö, J.; Valmu, L.; Salojärvi, J.; Palva, A.; Salonen, A.; de Vos, W.M. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE 2012, 7, e29913. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Giannone, R.J.; Morowitz, M.J.; Banfield, J.F.; Hettich, R.L. Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J. Proteom. Res. 2014, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Klaassens, E.S.; de Vos, W.M.; Vaughan, E.E. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl. Environ. Microbiol. 2007, 73, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2015, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of crohn’s disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [PubMed]
{kind=link}
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Licier, R.; Miranda, E.; Serrano, H. A Quantitative Proteomics Approach to Clinical Research with Non-Traditional Samples. Proteomes 2016, 4, 31. https://doi.org/10.3390/proteomes4040031
Licier R, Miranda E, Serrano H. A Quantitative Proteomics Approach to Clinical Research with Non-Traditional Samples. Proteomes. 2016; 4(4):31. https://doi.org/10.3390/proteomes4040031
Chicago/Turabian StyleLicier, Rígel, Eric Miranda, and Horacio Serrano. 2016. "A Quantitative Proteomics Approach to Clinical Research with Non-Traditional Samples" Proteomes 4, no. 4: 31. https://doi.org/10.3390/proteomes4040031
APA StyleLicier, R., Miranda, E., & Serrano, H. (2016). A Quantitative Proteomics Approach to Clinical Research with Non-Traditional Samples. Proteomes, 4(4), 31. https://doi.org/10.3390/proteomes4040031