
Spatial Proteomics for the Molecular Characterization of Breast Cancer

 ,
,  and
and
Abstract
1. Introduction
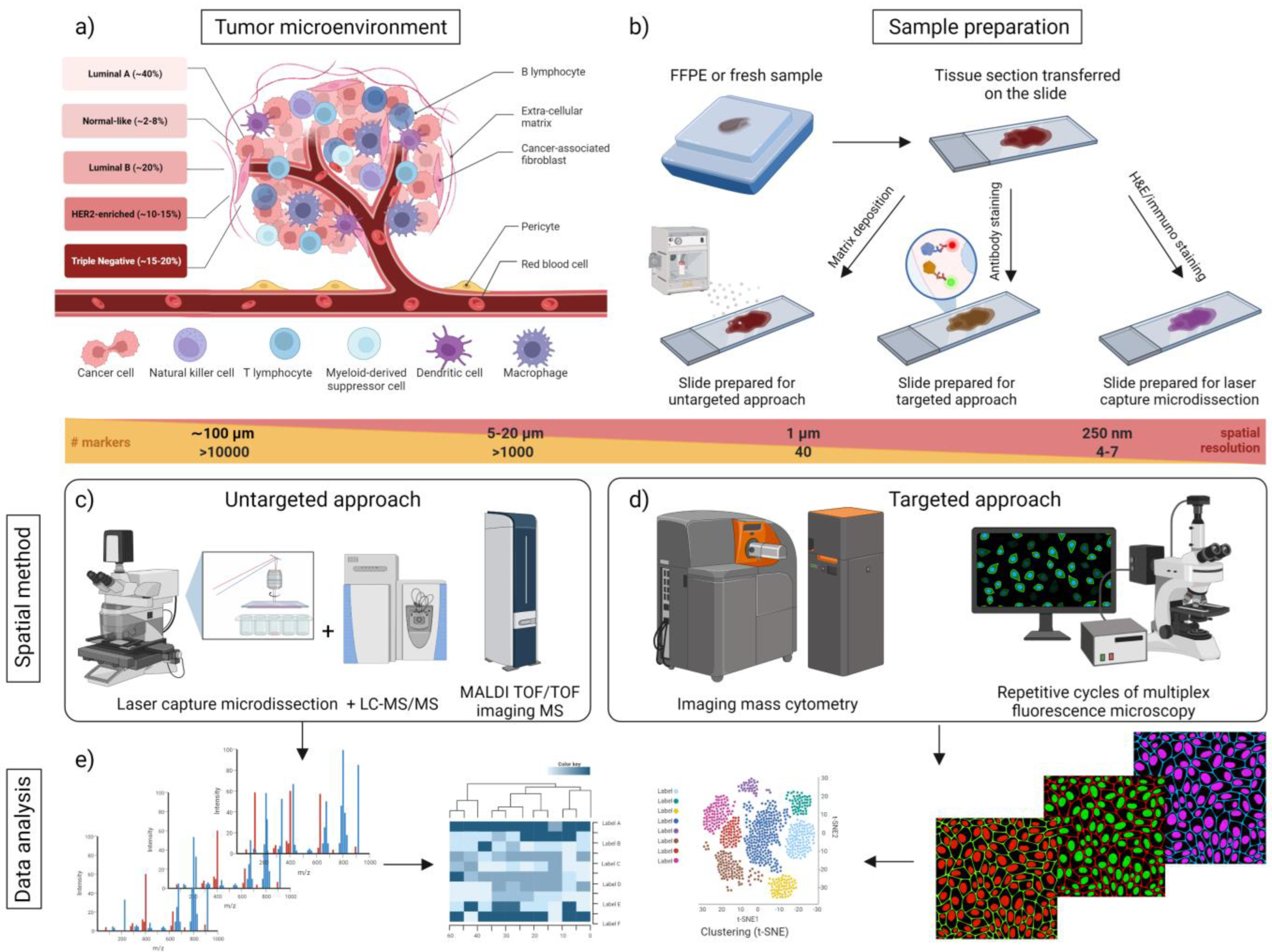
2. Breast Cancer Diagnosis and Classification
3. Molecular Biology of Breast Cancer
4. Proteomics Technologies for Spatial BC Analysis
4.1. Untargeted Spatial Proteomic Analysis (Untargeted MS and Imaging Mass Spectrometry)
4.1.1. Untargeted LC-MS for Spatial Proteomics
4.1.2. Imaging Mass Spectrometry (IMS)
4.2. Targeted Spatial Proteomic Analysis
4.2.1. Targeted Mass Spectrometry
4.2.2. Antibody-Based Spatial Proteomics (Imaging Techniques Using Single or Multiplexed Antibody Probes)
5. Concluding Remarks and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, G.; Alhaiek, A.A.; Godovac-Zimmermann, J. Proteomics reveals the importance of the dynamic redistribution of the subcellular location of proteins in breast cancer cells. Expert Rev. Proteom. 2015, 12, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Gnann, C.; Cesnik, A.J.; Lundberg, E. Illuminating Non-genetic Cellular Heterogeneity with Imaging-Based Spatial Proteomics. Trends Cancer 2021, 7, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Tainsky, M.A. Genomic and proteomic biomarkers for cancer: A multitude of opportunities. Biochim. Biophys. Acta 2009, 1796, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Duffy, M.J. Prognostic and predictive biomarkers in breast cancer: Past, present and future. Semin. Cancer Biol. 2018, 52, 56–73. [Google Scholar] [CrossRef]
- Riihimaki, M.; Thomsen, H.; Brandt, A.; Sundquist, J.; Hemminki, K. Death causes in breast cancer patients. Ann. Oncol. 2012, 23, 604–610. [Google Scholar] [CrossRef]
- Afzal, S.; Hassan, M.; Ullah, S.; Abbas, H.; Tawakkal, F.; Khan, M.A. Breast Cancer; Discovery of Novel Diagnostic Biomarkers, Drug Resistance, and Therapeutic Implications. Front. Mol. Biosci. 2022, 9, 783450. [Google Scholar] [CrossRef]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinari, S.; Moses, T.M.; Wong, S.; Dinh, J.; Christoforides, A.; Blum, J.L.; et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef]
- Visvader, J.E.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef]
- Granat, L.M.; Kambhampati, O.; Klosek, S.; Niedzwecki, B.; Parsa, K.; Zhang, D. The promises and challenges of patient-derived tumor organoids in drug development and precision oncology. Anim. Model. Exp. Med. 2019, 2, 150–161. [Google Scholar] [CrossRef]
- Grimwade, L.F.; Fuller, K.A.; Erber, W.N. Applications of imaging flow cytometry in the diagnostic assessment of acute leukaemia. Methods 2017, 112, 39–45. [Google Scholar] [CrossRef]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thurlimann, B.; Senn, H.J.; Panel, M. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Wolff, A.C.; Mangu, P.B.; Temin, S. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Oncol. Pract. 2010, 6, 195–197. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.S.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef]
- Pavlidis, N.; Pentheroudakis, G. Cancer of unknown primary site. Lancet 2012, 379, 1428–1435. [Google Scholar] [CrossRef]
- Varadhachary, G.R.; Spector, Y.; Abbruzzese, J.L.; Rosenwald, S.; Wang, H.; Aharonov, R.; Carlson, H.R.; Cohen, D.; Karanth, S.; Macinskas, J.; et al. Prospective gene signature study using microRNA to identify the tissue of origin in patients with carcinoma of unknown primary. Clin. Cancer Res. 2011, 17, 4063–4070. [Google Scholar] [CrossRef]
- Casadonte, R.; Kriegsmann, M.; Zweynert, F.; Friedrich, K.; Baretton, G.; Otto, M.; Deininger, S.O.; Paape, R.; Belau, E.; Suckau, D.; et al. Imaging mass spectrometry to discriminate breast from pancreatic cancer metastasis in formalin-fixed paraffin-embedded tissues. Proteomics 2014, 14, 956–964. [Google Scholar] [CrossRef]
- Duffy, M.J.; O'Donovan, N.; McDermott, E.; Crown, J. Validated biomarkers: The key to precision treatment in patients with breast cancer. Breast 2016, 29, 192–201. [Google Scholar] [CrossRef]
- Gamez-Pozo, A.; Trilla-Fuertes, L.; Berges-Soria, J.; Selevsek, N.; Lopez-Vacas, R.; Diaz-Almiron, M.; Nanni, P.; Arevalillo, J.M.; Navarro, H.; Grossmann, J.; et al. Functional proteomics outlines the complexity of breast cancer molecular subtypes. Sci. Rep. 2017, 7, 10100. [Google Scholar] [CrossRef]
- Loi, S.; Sirtaine, N.; Piette, F.; Salgado, R.; Viale, G.; Van Eenoo, F.; Rouas, G.; Francis, P.; Crown, J.P.; Hitre, E.; et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 2013, 31, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Muller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-Associated Lymphocytes As an Independent Predictor of Response to Neoadjuvant Chemotherapy in Breast Cancer. J. Clin. Oncol. 2010, 28, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Su, S.C.; Liu, Q.; Chen, J.Q.; Chen, J.N.; Chen, F.; He, C.H.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef]
- Wu, Y.; Cheng, Y.; Wang, X.; Fan, J.; Gao, Q. Spatial omics: Navigating to the golden era of cancer research. Clin. Transl. Med. 2022, 12, e696. [Google Scholar] [CrossRef]
- Pietras, R.J.; Arboleda, J.; Reese, D.M.; Wongvipat, N.; Pegram, M.D.; Ramos, L.; Gorman, C.M.; Parker, M.G.; Sliwkowski, M.X.; Slamon, D.J. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone-independent growth in human breast cancer cells. Oncogene 1995, 10, 2435–2446. [Google Scholar]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef]
- Song, D.; Cui, M.; Zhao, G.; Fan, Z.; Nolan, K.; Yang, Y.; Lee, P.; Ye, F.; Zhang, D.Y. Pathway-based analysis of breast cancer. Am. J. Transl. Res. 2014, 6, 302–311. [Google Scholar]
- Ortega, M.A.; Fraile-Martinez, O.; Asunsolo, A.; Bujan, J.; Garcia-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/mTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef]
- Schramm, G.; Kannabiran, N.; Konig, R. Regulation patterns in signaling networks of cancer. BMC Syst. Biol. 2010, 4, 162. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- El Ansari, R.; McIntyre, A.; Craze, M.L.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Altered glutamine metabolism in breast cancer; subtype dependencies and alternative adaptations. Histopathology 2018, 72, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, S.; Wang, X. The Metabolic Mechanisms of Breast Cancer Metastasis. Front. Oncol. 2020, 10, 602416. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zangar, R.C. Protein modifications as potential biomarkers in breast cancer. Biomark. Insights 2009, 4, 191–200. [Google Scholar] [CrossRef]
- Theivendran, S.; Tang, J.; Lei, C.; Yang, Y.; Song, H.; Gu, Z.; Wang, Y.; Yang, Y.; Jin, L.; Yu, C. Post translational modification-assisted cancer immunotherapy for effective breast cancer treatment. Chem. Sci. 2020, 11, 10421–10430. [Google Scholar] [CrossRef]
- Duong, V.; Bret, C.; Altucci, L.; Mai, A.; Duraffourd, C.; Loubersac, J.; Harmand, P.O.; Bonnet, S.; Valente, S.; Maudelonde, T.; et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol. Cancer Res. 2008, 6, 1908–1919. [Google Scholar] [CrossRef]
- Heo, K.S. Regulation of post-translational modification in breast cancer treatment. BMB Rep. 2019, 52, 113–118. [Google Scholar] [CrossRef]
- Rabellino, A.; Khanna, K.K. The implication of the SUMOylation pathway in breast cancer pathogenesis and treatment. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 54–70. [Google Scholar] [CrossRef]
- Zhai, Q.; Fan, J.; Lin, Q.; Liu, X.; Li, J.; Hong, R.; Wang, S. Tumor stromal type is associated with stromal PD-L1 expression and predicts outcomes in breast cancer. PLoS ONE 2019, 14, e0223325. [Google Scholar] [CrossRef]
- Boyages, J. Radiation therapy and early breast cancer: Current controversies. Med. J. Aust. 2017, 207, 216–222. [Google Scholar] [CrossRef]
- Goetz, J.G.; Minguet, S.; Navarro-Lerida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibanez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Berghoff, A.; Dinhof, C.; Jakesz, R.; Gnant, M.; Dubsky, P.; Jesch, B.; Heinzl, H.; Birner, P. Podoplanin-expressing cancer-associated fibroblasts are associated with poor prognosis in invasive breast cancer. Breast Cancer Res. Treat. 2012, 134, 237–244. [Google Scholar] [CrossRef]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; van den Brule, F.; Velculescu, V.; Polyak, K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef]
- Taylor, M.J.; Lukowski, J.K.; Anderton, C.R. Spatially Resolved Mass Spectrometry at the Single Cell: Recent Innovations in Proteomics and Metabolomics. J. Am. Soc. Mass. Spectr. 2021, 32, 872–894. [Google Scholar] [CrossRef]
- Irish, J.M.; Kotecha, N.; Nolan, G.P. Mapping normal and cancer cell signalling networks: Towards single-cell proteomics. Nat. Rev. Cancer 2006, 6, 146–155. [Google Scholar] [CrossRef]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug- induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O'Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef]
- Lee, H.J.; Jedrychowski, M.P.; Vinayagam, A.; Wu, N.; Shyh-Chang, N.; Hu, Y.; Min-Wen, C.; Moore, J.K.; Asara, J.M.; Lyssiotis, C.A.; et al. Proteomic and Metabolomic Characterization of a Mammalian Cellular Transition from Quiescence to Proliferation. Cell Rep. 2017, 20, 721–736. [Google Scholar] [CrossRef]
- Vistain, L.F.; Tay, S. Single-Cell Proteomics. Trends Biochem. Sci. 2021, 46, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Chang, J.T.; Echeverria, G.V. Methodological Advancements for Investigating Intra-tumoral Heterogeneity in Breast Cancer at the Bench and Bedside. J. Mammary Gland. Biol. 2020, 25, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Gromov, P.; Moreira, J.M.; Gromova, I. Proteomic analysis of tissue samples in translational breast cancer research. Expert. Rev. Proteom. 2014, 11, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Foss, E.J.; Radulovic, D.; Shaffer, S.A.; Goodlett, D.R.; Kruglyak, L.; Bedalov, A. Genetic Variation Shapes Protein Networks Mainly through Non-transcriptional Mechanisms. PLoS Biol. 2011, 9, e1001144. [Google Scholar] [CrossRef]
- Drissi, R.; Dubois, M.L.; Boisvert, F.M. Proteomics methods for subcellular proteome analysis. FEBS J. 2013, 280, 5626–5634. [Google Scholar] [CrossRef]
- Da Costa, G.G.; Gomig, T.H.; Kaviski, R.; Santos Sousa, K.; Kukolj, C.; De Lima, R.S.; De Andrade Urban, C.; Cavalli, I.J.; Ribeiro, E.M. Comparative Proteomics of Tumor and Paired Normal Breast Tissue Highlights Potential Biomarkers in Breast Cancer. Cancer Genom. Proteom. 2015, 12, 251–261. [Google Scholar]
- Meftahi, G.H.; Bahari, Z.; Zarei Mahmoudabadi, A.; Iman, M.; Jangravi, Z. Applications of western blot technique: From bench to bedside. Biochem. Mol. Biol. Educ. 2021, 49, 509–517. [Google Scholar] [CrossRef]
- Duncombe, T.A.; Kang, C.C.; Maity, S.; Ward, T.M.; Pegram, M.D.; Murthy, N.; Herr, A.E. Hydrogel Pore-Size Modulation for Enhanced Single-Cell Western Blotting. Adv. Mater. 2016, 28, 327–334. [Google Scholar] [CrossRef]
- Herzog, R.; Wagner, A.; Wrettos, G.; Stampf, K.; Bromberger, S.; Sperl, E.; Kratochwill, K. Improved Alignment and Quantification of Protein Signals in Two-Dimensional Western Blotting. J. Proteome Res. 2020, 19, 2379–2390. [Google Scholar] [CrossRef]
- Petrosius, V.; Schoof, E.M. Recent advances in the field of single-cell proteomics. Transl. Oncol. 2023, 27, 101556. [Google Scholar] [CrossRef]
- Lee, P.Y.; Yeoh, Y.; Omar, N.; Pung, Y.F.; Lim, L.C.; Low, T.Y. Molecular tissue profiling by MALDI imaging: Recent progress and applications in cancer research. Crit. Rev. Clin. Lab. Sci. 2021, 58, 513–529. [Google Scholar] [CrossRef]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef]
- Karas, M.; Bachmann, D.; Bahr, U.; Hillenkamp, F. Matrix-Assisted Ultraviolet-Laser Desorption of Nonvolatile Compounds. Int. J. Mass. Spectrom. 1987, 78, 53–68. [Google Scholar] [CrossRef]
- Mund, A.; Coscia, F.; Kriston, A.; Hollandi, R.; Kovacs, F.; Brunner, A.D.; Migh, E.; Schweizer, L.; Santos, A.; Bzorek, M.; et al. Deep Visual Proteomics defines single-cell identity and heterogeneity. Nat. Biotechnol. 2022, 40, 1231–1240. [Google Scholar] [CrossRef]
- Voskuil, J.L. The challenges with the validation of research antibodies. F1000Res 2017, 6, 161. [Google Scholar] [CrossRef]
- Gauthier, D.J.; Lazure, C. Complementary methods to assist subcellular fractionation in organellar proteomics. Expert. Rev. Proteom. 2008, 5, 603–617. [Google Scholar] [CrossRef]
- Sobsey, C.A.; Ibrahim, S.; Richard, V.R.; Gaspar, V.; Mitsa, G.; Lacasse, V.; Zahedi, R.P.; Batist, G.; Borchers, C.H. Targeted and Untargeted Proteomics Approaches in Biomarker Development. Proteomics 2020, 20, e1900029. [Google Scholar] [CrossRef]
- Crockett, D.K.; Lin, Z.; Vaughn, C.P.; Lim, M.S.; Elenitoba-Johnson, K.S. Identification of proteins from formalin-fixed paraffin-embedded cells by LC-MS/MS. Lab. Investig. 2005, 85, 1405–1415. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L.; Proteomics, C.T.D. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Curran, S.; McKay, J.A.; McLeod, H.L.; Murray, G.I. Laser capture microscopy. Mol. Pathol. 2000, 53, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Pappalardo, P.A.; Carpino, A.; Haymond, A.; Howard, M.; Espina, V.; Wulfkuhle, J.; Petricoin, E. Laser Capture Proteomics: Spatial tissue molecular profiling from the bench to personalized medicine. Expert. Rev. Proteom. 2021, 18, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Walch, A. Qualitative and quantitative mass spectrometry imaging of drugs and metabolites in tissue at therapeutic levels. Histochem. Cell Biol. 2013, 140, 93–104. [Google Scholar] [CrossRef]
- Lemaire, R.; Desmons, A.; Tabet, J.C.; Day, R.; Salzet, M.; Fournier, I. Direct analysis and MALDI imaging of formalin-fixed, paraffin-embedded tissue sections. J. Proteome Res. 2007, 6, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Kaletas, B.K.; van der Wiel, I.M.; Stauber, J.; Lennard, J.D.; Guzel, C.; Kros, J.M.; Luider, T.M.; Heeren, R.M. Sample preparation issues for tissue imaging by imaging MS. Proteomics 2009, 9, 2622–2633. [Google Scholar] [CrossRef]
- Schwartz, S.A.; Reyzer, M.L.; Caprioli, R.M. Direct tissue analysis using matrix-assisted laser desorption/ionization mass spectrometry: Practical aspects of sample preparation. J. Mass. Spectrom. 2003, 38, 699–708. [Google Scholar] [CrossRef]
- Kokkat, T.J.; Patel, M.S.; McGarvey, D.; LiVolsi, V.A.; Baloch, Z.W. Archived formalin-fixed paraffin-embedded (FFPE) blocks: A valuable underexploited resource for extraction of DNA, RNA, and protein. Biopreserv. Biobank 2013, 11, 101–106. [Google Scholar] [CrossRef]
- Nirmalan, N.J.; Harnden, P.; Selby, P.J.; Banks, R.E. Mining the archival formalin-fixed paraffin-embedded tissue proteome: Opportunities and challenges. Mol. Biosyst. 2008, 4, 712–720. [Google Scholar] [CrossRef]
- Gill, E.L.; Yost, R.A.; Vedam-Mai, V.; Garrett, T.J. Precast Gelatin-Based Molds for Tissue Embedding Compatible with Mass Spectrometry Imaging. Anal. Chem. 2017, 89, 576–580. [Google Scholar] [CrossRef]
- Nilsson, A.; Peric, A.; Strimfors, M.; Goodwin, R.J.A.; Hayes, M.A.; Andren, P.E.; Hilgendorf, C. Mass Spectrometry Imaging proves differential absorption profiles of well-characterised permeability markers along the crypt-villus axis. Sci. Rep. 2017, 7, 6352. [Google Scholar] [CrossRef]
- Carter, C.L.; Jones, J.W.; Farese, A.M.; MacVittie, T.J.; Kane, M.A. Inflation-Fixation Method for Lipidomic Mapping of Lung Biopsies by Matrix Assisted Laser Desorption/Ionization-Mass Spectrometry Imaging. Anal. Chem. 2016, 88, 4788–4794. [Google Scholar] [CrossRef]
- Norris, J.L.; Caprioli, R.M. Analysis of tissue specimens by matrix-assisted laser desorption/ionization imaging mass spectrometry in biological and clinical research. Chem. Rev. 2013, 113, 2309–2342. [Google Scholar] [CrossRef]
- Angel, P.M.; Comte-Walters, S.; Ball, L.E.; Talbot, K.; Mehta, A.; Brockbank, K.G.M.; Drake, R.R. Mapping Extracellular Matrix Proteins in Formalin-Fixed, Paraffin-Embedded Tissues by MALDI Imaging Mass Spectrometry. J. Proteome Res. 2018, 17, 635–646. [Google Scholar] [CrossRef]
- Hanton, S.D.; Clark, P.A.C.; Owens, K.G. Investigations of matrix-assisted laser desorption/ionization sample preparation by time-of-flight secondary ion mass spectrometry. J. Am. Soc. Mass. Spectr. 1999, 10, 104–111. [Google Scholar] [CrossRef]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef]
- Amstalden van Hove, E.R.; Smith, D.F.; Heeren, R.M. A concise review of mass spectrometry imaging. J. Chromatogr. A 2010, 1217, 3946–3954. [Google Scholar] [CrossRef]
- Chen, K.; Baluya, D.; Tosun, M.; Li, F.; Maletic-Savatic, M. Imaging Mass Spectrometry: A New Tool to Assess Molecular Underpinnings of Neurodegeneration. Metabolites 2019, 9, 135. [Google Scholar] [CrossRef]
- Glish, G.L.; Vachet, R.W. The basics of mass spectrometry in the twenty-first century. Nat. Rev. Drug. Discov. 2003, 2, 140–150. [Google Scholar] [CrossRef]
- Dong, Y.H.; Aharoni, A. Image to insight: Exploring natural products through mass spectrometry imaging. Nat. Prod. Rep. 2022, 39, 1510–1530. [Google Scholar] [CrossRef]
- Hajjaji, N.; Abbouchi, M.; Nguyen, L.A.; Charles, S.; Leclercq, S.; Bertin, D.; Robin, Y.-M.; Fournier, I.; Salzet, M. A novel proteomic mass spectrometry-based approach to reveal functionally heterogeneous tumor clones in breast cancer metastases and identify clone-specific drug targets. J. Clin. Oncol. 2020, 38, e13063. [Google Scholar] [CrossRef]
- Stauber, J.; MacAleese, L.; Franck, J.; Claude, E.; Snel, M.; Kaletas, B.K.; Wiel, I.M.; Wisztorski, M.; Fournier, I.; Heeren, R.M. On-tissue protein identification and imaging by MALDI-ion mobility mass spectrometry. J. Am. Soc. Mass. Spectrom. 2010, 21, 338–347. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, L.; Li, R. HnRNPA2/B1 Is a Novel Prognostic Biomarker for Breast Cancer Patients. Genet. Test. Mol. Biomark. 2020, 24, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Ouban, A. Filamin-A expression in triple-negative breast cancer and its clinical significance. Biotechnol. Biotechnol. Equip. 2021, 35, 1409–1419. [Google Scholar] [CrossRef]
- Soltwisch, J.; Kettling, H.; Vens-Cappell, S.; Wiegelmann, M.; Muthing, J.; Dreisewerd, K. Mass spectrometry imaging with laser-induced postionization. Science 2015, 348, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Zavalin, A.; Todd, E.M.; Rawhouser, P.D.; Yang, J.H.; Norris, J.L.; Caprioli, R.M. Direct imaging of single cells and tissue at sub-cellular spatial resolution using transmission geometry MALDI MS. J. Mass Spectrom. 2012, 47, 1473–1481. [Google Scholar] [CrossRef]
- Niehaus, M.; Soltwisch, J.; Belov, M.E.; Dreisewerd, K. Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at subcellular resolution. Nat. Methods 2019, 16, 925–931. [Google Scholar] [CrossRef]
- Jung, J. Recent Advances of MALDI-Mass Spectrometry Imaging in Cancer Research. Mass Spectrom. Lett. 2019, 10, 71–78. [Google Scholar] [CrossRef]
- Manes, N.P.; Nita-Lazar, A. Application of targeted mass spectrometry in bottom-up proteomics for systems biology research. J. Proteom. 2018, 189, 75–90. [Google Scholar] [CrossRef]
- Shi, T.; Song, E.; Nie, S.; Rodland, K.D.; Liu, T.; Qian, W.J.; Smith, R.D. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016, 16, 2160–2182. [Google Scholar] [CrossRef]
- Murray, K.K.; Boyd, R.K.; Eberlin, M.N.; Langley, G.J.; Li, L.; Naito, Y. Definitions of terms relating to mass spectrometry (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1515–1609. [Google Scholar] [CrossRef]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef]
- McLuckey, S.A. Principles of collisional activation in analytical mass spectrometry. J. Am. Soc. Mass Spectrom. 1992, 3, 599–614. [Google Scholar] [CrossRef]
- Dongre, A.R.; Somogyi, A.; Wysocki, V.H. Surface-induced dissociation: An effective tool to probe structure, energetics and fragmentation mechanisms of protonated peptides. J. Mass Spectrom. 1996, 31, 339–350. [Google Scholar] [CrossRef]
- Kim, M.S.; Pandey, A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics 2012, 12, 530–542. [Google Scholar] [CrossRef]
- Steiner, C.; Lescuyer, P.; Cutler, P.; Tille, J.C.; Ducret, A. Relative Quantification of Proteins in Formalin-Fixed Paraffin-Embedded Breast Cancer Tissue Using Multiplexed Mass Spectrometry Assays. Mol. Cell Proteomics 2022, 21, 100416. [Google Scholar] [CrossRef]
- Badve, S.S.; Gokmen-Polar, Y. Protein Profiling of Breast Cancer for Treatment Decision-Making. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 73–81. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef]
- Yamauchi, K.A.; Herr, A.E. Subcellular western blotting of single cells. Microsyst. Nanoeng. 2017, 3, 16079. [Google Scholar] [CrossRef]
- Im, K.; Mareninov, S.; Diaz, M.F.P.; Yong, W.H. An Introduction to Performing Immunofluorescence Staining. Methods Mol. Biol. 2019, 1897, 299–311. [Google Scholar] [CrossRef]
- Furia, L.; Pelicci, S.; Perillo, F.; Bolognesi, M.M.; Pelicci, P.G.; Facciotti, F.; Cattoretti, G.; Faretta, M. Automated multimodal fluorescence microscopy for hyperplex spatial-proteomics: Coupling microfluidic-based immunofluorescence to high resolution, high sensitivity, three-dimensional analysis of histological slides. Front. Oncol. 2022, 12, 960734. [Google Scholar] [CrossRef]
- Allam, M.; Cai, S.; Coskun, A.F. Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics. NPJ Precis. Oncol. 2020, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, M.J.; Sevinsky, C.J.; Sood, A.; Adak, S.; Bello, M.O.; Bordwell, A.; Can, A.; Corwin, A.; Dinn, S.; Filkins, R.J.; et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. USA 2013, 110, 11982–11987. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Izar, B.; Wang, S.; Yapp, C.; Mei, S.; Shah, P.M.; Santagata, S.; Sorger, P.K. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 2018, 7, e31657. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, M.J.; Gokmen-Polar, Y.; Sui, Y.; Pang, A.S.; LaPlante, N.; Harris, A.L.; Tan, P.H.; Ginty, F.; Badve, S.S. Single-cell heterogeneity in ductal carcinoma in situ of breast. Mod. Pathol. 2018, 31, 406–417. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981.e915. [Google Scholar] [CrossRef]
- Mishra, S.; Charan, M.; Shukla, R.K.; Agarwal, P.; Misri, S.; Verma, A.K.; Ahirwar, D.K.; Siddiqui, J.; Kaul, K.; Sahu, N.; et al. cPLA2 blockade attenuates S100A7-mediated breast tumorigenicity by inhibiting the immunosuppressive tumor microenvironment. J. Exp. Clin. Cancer Res. 2022, 41, 54. [Google Scholar] [CrossRef]
- Magaki, S.; Hojat, S.A.; Wei, B.; So, A.; Yong, W.H. An Introduction to the Performance of Immunohistochemistry. Methods Mol. Biol. 2019, 1897, 289–298. [Google Scholar] [CrossRef]
- Rojo, F.; Gonzalez-Perez, A.; Furriol, J.; Nicolau, M.J.; Ferrer, J.; Burgues, O.; Sabbaghi, M.; Gonzalez-Navarrete, I.; Cristobal, I.; Serrano, L.; et al. Non-canonical NF-kappaB pathway activation predicts outcome in borderline oestrogen receptor positive breast carcinoma. Br. J. Cancer 2016, 115, 322–331. [Google Scholar] [CrossRef]
- Kinkhabwala, A.; Herbel, C.; Pankratz, J.; Yushchenko, D.A.; Ruberg, S.; Praveen, P.; Reiss, S.; Rodriguez, F.C.; Schafer, D.; Kollet, J.; et al. MACSima imaging cyclic staining (MICS) technology reveals combinatorial target pairs for CAR T cell treatment of solid tumors. Sci. Rep. 2022, 12, 1911. [Google Scholar] [CrossRef]
- Van Acker, T.; Buckle, T.; Van Malderen, S.J.M.; van Willigen, D.M.; van Unen, V.; van Leeuwen, F.W.B.; Vanhaecke, F. High-resolution imaging and single-cell analysis via laser ablation-inductively coupled plasma-mass spectrometry for the determination of membranous receptor expression levels in breast cancer cell lines using receptor-specific hybrid tracers. Anal. Chim. Acta 2019, 1074, 43–53. [Google Scholar] [CrossRef]
- Chang, Q.; Ornatsky, O.I.; Siddiqui, I.; Loboda, A.; Baranov, V.I.; Hedley, D.W. Imaging Mass Cytometry. Cytometry A 2017, 91, 160–169. [Google Scholar] [CrossRef]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- McDonnell, L.A.; Heeren, R.M. Imaging mass spectrometry. Mass Spectrom. Rev. 2007, 26, 606–643. [Google Scholar] [CrossRef]
- Ali, H.R.; Jackson, H.W.; Zanotelli, V.R.T.; Danenberg, E.; Fischer, J.R.; Bardwell, H.; Provenzano, E.; Team, C.I.G.C.; Rueda, O.M.; Chin, S.F.; et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 2020, 1, 163–175. [Google Scholar] [CrossRef]
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- McCart Reed, A.E.; Bennett, J.; Kutasovic, J.R.; Kalaw, E.; Ferguson, K.; Yeong, J.; Simpson, P.T.; Lakhani, S.R. Digital spatial profiling application in breast cancer: A user's perspective. Virchows Arch. 2020, 477, 885–890. [Google Scholar] [CrossRef]
- Yagnik, G.; Liu, Z.; Rothschild, K.J.; Lim, M.J. Highly Multiplexed Immunohistochemical MALDI-MS Imaging of Biomarkers in Tissues. J. Am. Soc. Mass Spectrom. 2021, 32, 977–988. [Google Scholar] [CrossRef]
- Alexandrov, T. MALDI imaging mass spectrometry: Statistical data analysis and current computational challenges. BMC Bioinform. 2012, 13 (Suppl. S16), S11. [Google Scholar] [CrossRef]
- Rafols, P.; Vilalta, D.; Brezmes, J.; Canellas, N.; Del Castillo, E.; Yanes, O.; Ramirez, N.; Correig, X. Signal preprocessing, multivariate analysis and software tools for MA(LDI)-TOF mass spectrometry imaging for biological applications. Mass Spectrom. Rev. 2018, 37, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Thiele, H.; Heldmann, S.; Trede, D.; Strehlow, J.; Wirtz, S.; Dreher, W.; Berger, J.; Oetjen, J.; Kobarg, J.H.; Fischer, B.; et al. 2D and 3D MALDI-imaging: Conceptual strategies for visualization and data mining. Biochim. Biophys Acta 2014, 1844, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Claesen, M.; Moerman, T.; Groseclose, M.R.; Waelkens, E.; De Moor, B.; Verbeeck, N. Spatially aware clustering of ion images in mass spectrometry imaging data using deep learning. Anal. Bioanal. Chem. 2021, 413, 2803–2819. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type | Spatial Method | Principle | Spatial Resolution | Multiplexing | Advantage | Disadvantage |
|---|---|---|---|---|---|---|
| Targeted | IF | Antibodies designed to target specific proteins | 250 nm | 1–5 | Signal amplification Resolution Analytical capabilities | Background signal and spectral overlap |
| Cell DIVE | Antibodies with cyclic oligo-barcoded reporter | 1 µm | >60 | Standardized workflows with automation | Restricted to regions of interestPotential for epitope loss | |
| CODEX | Antibodies with cyclic oligo-barcoded reporter | 1 µm | >60 | Standardized workflows with automation | Restricted to regions of interest | |
| MCI | Combination of metal-labeled antibody immunostaining and ultraviolet laser ablation | 1 µm | 40 | Minimal overlap or signal background | Requirement for expensive instrumentation and metal isotope-labeled antibodies | |
| MIBI | Combination of metal-labeled antibody immunostaining and ion-beam gun ablation | 1 µm | 40–100 | Minimal overlap or signalbackground | Requirement for expensive instrumentation and metal isotope-labeled antibodies | |
| MICS | Photobleaching of fluorescent labels of recombinant antibodies and release of antibodies or their labels | 1 µm | >100 | Compatible with other technologies | Duration of experiment | |
| DSP | UV-cleaved oligo-conjugated primary antibody and barcode counting | 5 µm | 90 | High multiplexing ability Non-destructive procedure | Restricted to regions of interest | |
| MALDI-IHC | Targeted IMS in combination with IHC | 5–10 µm | 12 | Nondestructive method No cyclic workflows required | Extra preparation steps Limited sensitivity High acquisition time | |
| Untargeted | t-MALDI-2 | Laser-induced post-ionization technique in transmission-mode geometry | <1 µm | >100 | Label-free conditions Compatible with subsequent H&E staining | Extra preparation steps Vacuum condition High acquisition time |
| MALDI-IMS | Ionization of all molecules within the pixel, generating a separate spectra per pixel | 5–20 µm | >1000 | Label-free conditions Compatible with subsequent H&E staining | Extra preparation steps Vacuum conditionLimit of detection High acquisition time | |
| LCM + LC-MS/MS | Isolation of specific cells on a tissue section using laser | 100 µm | >10,000 | Label-free conditions Ability to isolate specific cell types from heterogeneous tissues | Extra preparation steps Need for a pathologist |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brožová, K.; Hantusch, B.; Kenner, L.; Kratochwill, K. Spatial Proteomics for the Molecular Characterization of Breast Cancer. Proteomes 2023, 11, 17. https://doi.org/10.3390/proteomes11020017
Brožová K, Hantusch B, Kenner L, Kratochwill K. Spatial Proteomics for the Molecular Characterization of Breast Cancer. Proteomes. 2023; 11(2):17. https://doi.org/10.3390/proteomes11020017
Chicago/Turabian StyleBrožová, Klára, Brigitte Hantusch, Lukas Kenner, and Klaus Kratochwill. 2023. "Spatial Proteomics for the Molecular Characterization of Breast Cancer" Proteomes 11, no. 2: 17. https://doi.org/10.3390/proteomes11020017
APA StyleBrožová, K., Hantusch, B., Kenner, L., & Kratochwill, K. (2023). Spatial Proteomics for the Molecular Characterization of Breast Cancer. Proteomes, 11(2), 17. https://doi.org/10.3390/proteomes11020017