Abstract
Corynebacterium pseudotuberculosis is an important animal pathogen, which is also able to infect humans. An optimal treatment of infections with this pathogen is not available today and consequently, more research is necessary to understand the infection process. Here, we present a combined -omics and bioinformatics approach to characterize C. pseudotuberculosis 12CS0282. The genome sequence of strain 12CS0282 was determined, analyzed in comparison with the available 130 C. pseudotuberculosis sequences and used as a basis for proteome analyses. In a reverse vaccinology approach, putative vaccine and drug targets for 12CS0208 were identified. Mass spectrometry analyses revealed the presence of multiple virulence factors even without host contact. In macrophage interaction studies, C. pseudotuberculosis 12CS0282 was highly resistant against human phagocytes and even multiplied within human THP-1 cells. Taken together, the data indicate a high pathogenic potential of the strain.
1. Introduction
Corynebacterium pseudotuberculosis is a zoonotic pathogen closely related to Corynebacterium ulcerans and Corynebacterium diphtheriae. Based on the fact that these three species can be lsogenized by a corynebacteriophage [1], and as a result may carry the tox gene coding for diphtheria toxin (DT), C. pseudotuberculosis, C. ulcerans and C. diphtheriae are forming the group of toxigenic corynebacteria [2]. The species is divided into two biovars based on biochemical properties (e.g., nitrate metabolism), infected host animals and evoked diseases [3]. Biovar ovis is the causative agent of caseous lymphadenitis in small ruminants such as sheep and goats and mastitis in dairy cattle [4,5,6], while biovar equi causes abscesses as well as ulcerative lymphangitis in equines and oedematous skin disease in buffalos [7].
Due to the importance of host animals for milk, meat, leather and wool production, and the worldwide distribution of the pathogen, significant economic losses are resulting from C. pseudotuberculosis infections [3]. Furthermore, as a zoonotic pathogen, humans can also be infected with C. pseudotuberculosis causing lymphadenitis [8,9]. Consequently, it is not astonishing that a number of C. pseudotuberculosis sequencing and proteome projects have been carried out, beginning in 2010 (e.g., [9,10,11,12]), and often with the aim of identifying candidates for vaccine production (e.g., [13,14,15]). Nevertheless, as concluded in a recent analysis of transcriptional regulatory networks in C. pseudotuberculosis [16], more experimental studies are needed for a better understanding of this important animal pathogen.
Here, we present a combined genome and proteome analysis of C. pseudotuberculosis strain 12CS0208 isolated from a goat in Thuringia, Germany.
2. Materials and Methods
2.1. Bacteria and Growth Conditions
C. pseudotuberculosis strains 12CS0282 and FRC41, C. ulcerans 809, Corynebacterium glutamicum ATCC13032 and Corynebacterium silvaticum W25 were cultivated at 37 °C on Columbia Blood agar plates (CBA, Oxoid, Wesel, Germany) or in Brain Heart Infusion (BHI, Oxoid, Wesel, Germany) under constant shaking at 125 rpm in baffled flasks.
2.2. Genome Sequencing and Analysis
DNA for whole genome sequencing was prepared after cultivation in BHI using QIAGEN Genomic-tips 20/G and a QIAGEN Genomic DNA buffer set kit (Qiagen, Hilden, Germany). DNA was sequenced on an Illumina MiSeq instrument (Illumina, San Diego, CA, USA).
The paired-end sequencing reads were assembled using SPAdes 3.13.1 [17] and scaffolded using MeDuSa [18] with the genome sequence of C. pseudotuberculosis strain 29,156 used as the reference. The genome sequences of strains C. pseudotuberculosis 12CS0282 has been submitted to GenBank (Accession no.: NZ_CP074370.1).
16S rRNA gene sequence from the genome was extracted using RNAMMER/1.2 [19] and were aligned to 16S rRNA reference sequences for the genus Corynebacterium obtained from the GenBank using MUSCLE [20]. A maximum-likelihood tree was constructed from the alignment after removing the sites with the missing data using IQ-Tree following the GTR + F + I + G4 substitution model with 100,000 SH-aLRT and 100,000 ultrafast bootstrap iterations [21]. The tree was visualized using iTol server [22].
Digital DNA-DNA hybridization (dDDH) values were calculated between the genome sequence of C. pseudotuberculosis 12CS0282 and Corynebacterium belfantii DSM 105776 T (OANN01), C. diphtheriae DSM 44123 T (LJXR01), C. pseudotuberculosis ATCC19410 T (CP021251.1), C. pseudotuberculosis DSM 20689 T (RBXH01), C. silvaticum KL0182 T (SDQO02), C. silvaticum W25 (VFEM01), C. ulcerans FRC11 (CP009622.1) and C. ulcerans NCTC 7910 T (LT906443.1) using the GGDC server [23,24].
To compare the genomic variation with proteomic analyses, we obtained sequences of strains C. pseudotuberculosis 258 (GCA_000263755.3), C. silvaticum W25 (GCA_006370535.1), C. diphtheriae C7 (beta; GCA_000255175.1), C. ulcerans 809 (GCA_000215645.1), C. pseudotuberculosis 1/06A (GCA_000233735.1), C. pseudotuberculosis E16 (GCA_008461845.1), C. pseudotuberculosis I19 (GCA_000152065.3) and C. pseudotuberculosis FRC41 (GCA_000143705.2) from the Genbank. All genomes were annotated using Prokka [25] and were compared using Roary with a sequence identity cut-off of 70% [26,27]. Finally, all available 130 C. pseudotuberculosis genomes were also obtained from the GenBank and were compared as mentioned above.
The virulence genes previously reported in C. pseudotuberculosis strains [9] were searched across all C. pseudotuberculosis genomes using protein BLAST searches [28].
2.3. Sample Preparation of Proteomic Analyses
Preparation of whole cell proteins was carried out as described previously [29]. For this purpose, the cells were resuspended in PBS buffer with protease inhibitor (Complete, Roche, Basel, Switzerland) and lysed with a homogenizer (MP Biomedical, Fisher Scientific, Schwerte, Germany) using glass beads (5.5 m s−1, 30 s, 5 cycles, 4 °C). Cell debris was removed by centrifugation at 13,000× g for 30 min at 4 °C and the supernatant was retained. Protein buffer was added to a final concentration of 10 mM DTT, 2% sodium deoxycholate and 50 mM Tris (pH 8.0). For in solution tryptic digest, 40 µg of protein was transferred into a new low-binding tube and incubated at 55 °C for 30 min for protein denaturation followed by an alkylation of sulfhydryl groups (40 mM CAA final concentration). Precipitation of proteins was carried out overnight at 4 °C with 80% acetone, washed three times with 80% ice-cold acetone, dried under N2 atmosphere to avoid oxidation [30] and resuspended in 100 mM TEAB buffer. Proteins were digested overnight at 37 °C under constant shaking with 2 µg of trypsin. Desalting occurred with C18 stage tips with 25 µg of all peptide samples (three biological replicates of secreted proteins, whole proteome and surface proteins). Peptides were vacuum-dried and resuspended in 0.1% trifluoroacetic acid (TFA) for LC-MS/MS analysis [31].
For preparation of extracellular proteins, the bacteria were removed by centrifugation (10 min, 4 °C, 4000× g) and the supernatant was subsequently filtered using a 0.2 µm pore size filter (Minisart, Sartorius, Göttingen, Germany). The proteins were precipitated using 10% (w/v) trichloroacetic acid (TCA) and resuspended in protein buffer (10 mM DTT, 2% sodium deoxycholate, 50 mM Tris, pH 8.0). PierceTM 660 nm Protein Assay (Thermo Fisher Scientific, Bremen, Germany) was used to determine the protein concentration. 25 µg of the proteins were used for a tryptic digest on 10 kDa vivacon 500 membrane filters as previously described following a modified Filter Aided Sample preparation (FASP) protocol [30,32,33]. Cell surface proteins were isolated by tryptic shaving [31,34]. For this purpose, cells were harvested and treated with 1.5 µg sequencing grade trypsin (Promega, Madison, WI, USA) for 1.5 h at 37 °C.
2.4. Mass Spectrometry
Mass spectrometric analyses were carried as described before [29,30,31,33]. The separation of 10 µg of peptides were carried out by a nanoflow Ultimate 3000 HPLC (Dionex, Sunnyvale, CA, USA) using an EASY-Spray column (Thermo Fisher Scientific; C18 with 2 µm particle size, 50 cm × 75 µm) with a (flow rate of 200 nL min−1 and increasing acetonitrile concentrations over 120 min. The total method duration including equilibration and column wash was set to 160 min. Triplicates of all samples were analyzed using an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) with following settings: spray voltage 2000 V, transfer tube temperature 275 °C, scan range for the MS 1 detection in the Orbitrap 300–2000 (m/z), 50 ms maximum injection time, automatic gain control (AGC) target of 4 × 106 and Orbitrap resolution of 120.000 [29]. For collision-induced dissociation with a collision energy of 35%, the 10 most intense ions were selected and for ion trap detection a maximum injection time of 250 ms and an AGC target of 1 × 105 were set. Resulting raw data files were analyzed using the Proteome Discoverer 1.4 program package (Thermo Fisher Scientific, Bremen, Germany) and the theoretical proteome of C. pseudotuberculosis 12CS0282. As described by Schäfer and co-workers [35], the theoretical masses of peptides were generated with a maximum of two missed cleavages. Carbamidomethyl modification on cysteine was set as fixed modification, oxidation of methionine as dynamic modification. To compare the measured spectra of product ions, the mass tolerance for survey scans was set to 10 ppm and 0.6 Da for fragment mass measurements. False discovery rate (FDR) was set on 1% for protein identification.
2.5. Characterization and Visualization of Proteome Data
Localization prediction and detection of signal peptides was carried out using the psortb server [36] and LipoP [37]. Proteins including transmembrane helices were detected with the TMH MM Server v.2.0 tool [38,39]. For pathway analysis, amino acid sequences from the theoretical proteome were used for a BlastKOALA search [40]. Missing information was completed by a proteome comparison, carried out using the PATRIC 3.6.5 database [41] and extracted from existing data of homologous proteins from other C. pseudotuberculosis strains. Visualization of the generated data was carried out using the proteomaps program [42,43,44].
2.6. Reverse Vaccinology
For the identification of novel vaccine targets, an in silico reverse vaccinology approach was applied [45,46]. Surface associated and secreted proteins from the core proteome of C. pseudotuberculosis were extracted and analyzed for a putative essential function using the DEG 10 database [47]. Only proteins with the following parameters were considered as essential: bit score > 100, E-value with a cut-off of 1 × 10−4 and percentage of identity > 35% [48]. The remaining proteins were analyzed as probable vaccine targets and non-host-homologous proteins (human, pig and mouse) were excluded using the Vaxign2 prediction tool [49]. The antigen classification of the proteins was analyzed by VaxiJen [50]. For antigenicity analysis, the default parameter value was set to 0.4. The ProtParam tool [51] estimated the molecular weight (MW) and stability of the proteins (Figure 1).
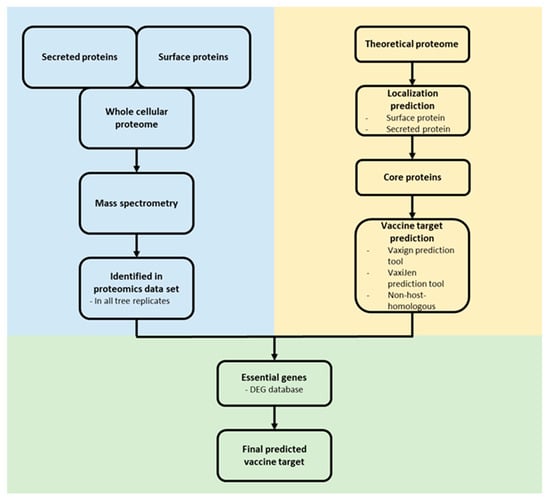
Figure 1.
Reverse vaccinology approach. Proteome data (blue) and in silico protein characterization (yellow) were combined to predict potential vaccine and drug targets (green).
To visualize protein-protein interaction networks of selected vaccine candidates from the reverse vaccinology approach the Search Tool for the Retrieval of Interacting Genes/Proteins–STRING was used with strain C231 as reference proteome [52].
2.7. Interaction of C. Pseudotuberculosis Strain 12CS0282 with Human Macrophages
Gentamicin protection assays were performed for quantitative analysis of invasion and survival of C. ulcerans 809, C. silvaticum W25, C. pseudotuberculosis 12CS0282 and C. pseudotuberculosis FRC41 in phagocytes. For this purpose, THP-1 cells, a human leukemia monocyte cell line, were differentiated by addition of 10 ng ml−1 phorbol 12-myristate 13-acetate. Cells were seeded in a density of 2 × 105 cells per well with 500 μL medium without antibiotics 24 h prior to infection in 24-well plates. Bacteria used for infection of the cells were inoculated from overnight cultures to an optical density at 600 nm (OD600) of 0.2 in fresh medium and incubated at 37 °C and 125 rpm until an OD600 of 0.4–0.6 was reached. Cells were harvested by centrifugation (4500× g, 5 min, 4 °C) and washed with cold PBS under the same conditions. The cell pellet was resuspended in 500 μL cold PBS and the OD600 was determined in triplicates and adjusted to a value of 1 in 1000 μL PBS and finally diluted 10−2. 50 μL of this cell suspension in 450 μL prewarmed cell culture medium without antibiotics per well were used as inoculum which results in a multiplicity of infection (MOI) of 1. Serial dilutions of the inoculi were plated on blood agar plates (Oxoid, Wesel, Germany) using an Eddy Jet Version 1.22 (IUL Instruments, Barcelona, Spain) and incubated at 37 °C for two days. The infection plates were centrifuged for 5 min at 350× g to synchronize the infection and incubated at cell culture conditions for 30 min to allow phagocytosis of bacteria. Subsequently, the supernatant containing non-engulfed bacteria was aspirated, cells were washed once with PBS and remaining extracellular bacteria were killed by addition of 100 μg mL−1 gentamicin in cell culture medium. After 2 h, cells were either lysed by adding 500 μL of 0.1% Triton X-100 in PBS and intracellular bacteria were recovered by plating serial dilutions of the lysates on blood agar plates or further incubated for analysis at later time points (4 and 20 h). After incubation at 37 °C for two days, the number of colony forming units (CFU) was determined. The ratio of bacteria used for infection (number on colonies on the inoculum plates) and bacteria on the lysate (number of colonies on the lysate plates) multiplied with 100 gave the percentage of viable intracellular bacteria at different time points. When the survival of intracellular bacteria in THP-1 cells was analyzed over the time, the number of CFU at 2 h was set to 100% and later time points were calculated based on this value.
2.8. Statistical Considerations
For label-free quantification only proteins, which are present in all three independent biological replicates, were considered as identified. The peak areas of the identified proteins were normalized via the molecular weight and the relative abundance of each protein was calculated based on the total protein approach (TPA) method [29].
For gentamicin protection assays, experiments were performed in three independent biological replicates with technical triplicates (n = 9) and means and standard deviations were calculated. Unpaired Student’s t-test was performed using GraphPad Prism 7.0 (GraphPad, San Diego, CA, USA).
3. Results
3.1. Genome Analysis
3.1.1. Phylogenomic Characteristics of Strain 12CS0282
The genome sequence of strain 12CS0282 is 2.3 Mb in size with a GC content of 52.2 mol%. 2119 coding sequences, 5 rRNA, 49 tRNA and 1 tmRNA were annotated.
The phylogenetic tree calculated from the 16S rRNA sequences grouped strain 12CS0282 with other C. pseudotuberculosis strains (Figure 2). To further confirm the strain identity, we calculated digital DNA-DNA hybridization values between strain 12CS0282 and representatives of closely related corynebacterial species. The program GGDC 3.0 uses three approaches (formulas) for calculation of the dDDH values, but the values derived from formula #2 are recommended for the dataset that includes draft assemblies [23,53]. These values are summarized in Table 1. dDDH values between strain 12CS0282 and the type strain of C. pseudotuberculosis were above the 70% cut-off value to define species, confirming that strain 12CS0282 belongs to C. pseudotuberculosis. The dDDH values between this strain and other Corynebacterium species was <30% (Table 1).
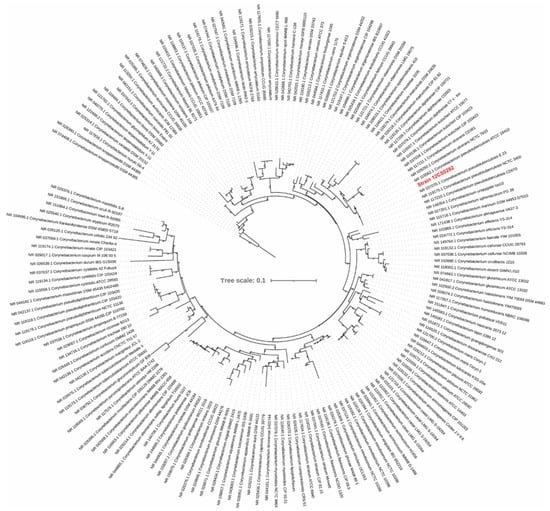
Figure 2.
A maximum-likelihood tree from the 16S rRNA gene sequence alignment. The scale bar represents nucleotide substitution per site. Strain 12CS0282 is highlighted in red.

Table 1.
Digital DNA-DNA hybridization values between strain 12CS0282 and representative strains of closely related species.
A comparative analysis of 131 genomes indicated that approximately 2/3 of C. pseudotuberculosis genome is highly conserved genome with 1562 genes present in 99–100% strains. The size of the pan-genome is 3778 genes including 250 Soft core genes (95–99% strains), 620 Shell genes (15–95% strains) and 1346 Cloud genes (0- 15% strains). A phylogenetic tree from the core genome separated C. pseudotuberculosis strains into two major clades (Figure 3 and Figure S1). This is consistent with the previous studies [12,13]. Clade 1 include 95 strains including all strains identified as biovar ovis (Supplementary Figure S1). 33 strains grouped in Clade 2 and include most of the strains identified as biovar equi (Supplementary Figure S1). Three strains were quite distinct from both the clades, including one strain, Cp162, also identified as biovar equi. These observations are consistent with the findings of Soares and coworker [13].
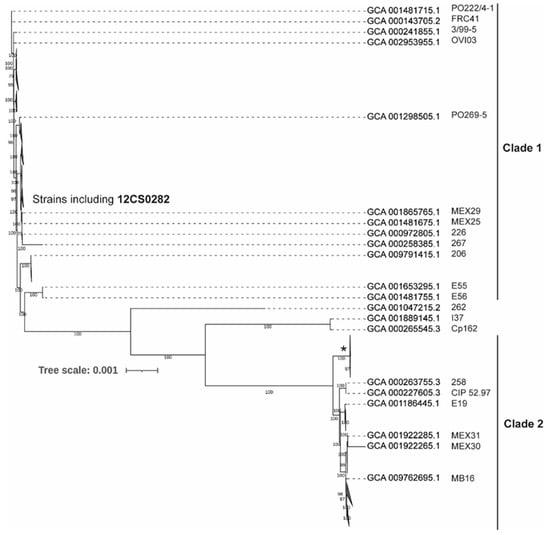
Figure 3.
Maximum-likelihood tree from nucleotide sequence alignment of the core genome calculated from 131 C. pseudotuberculosis strains. Clades with the average branch distance to their leaves below 0.0001 were collapsed. Nine out of 11 strains in the clade marked by * possess the tox gene. The scale bar represents nucleotide substitution per site (see Supplementary Figure S1 for the all strain details).
3.1.2. Virulence Genes in Strain 12CS0282
A protein BLAST search for the pilus gene clusters identified both spaBC (cp12CS0282_00823-cp12CS0282_00828) and spaDEF (cp12CS0282_00855- cp12CS0282_00860) type cluster in strain 12CS0282. However, cp12CS0282_00826 to cp12CS0282_00828 are partial SpaC sequences. Therefore, spaC is a pseudogene in this strain. The SpaDEF type cluster has all required genes in this strain [9]. Other virulence genes reported in C. pseudotuberculosis are also present in the strain 12CS0282 (Table 2), suggesting that this strain is well equipped to cause infection in sheep and other animals. The tox gene encoding diphtheria-like toxin is absent in this strain.
Table 2.
Presence of known virulence genes in strain 12CS0282.
We also searched for the presence of these proteins among other 130 C. pseudotuberculosis strains and found that most of them are highly conserved across the dataset with minor exceptions (Supplementary Table S1). Corynebacterial protease CP40 (Cpp) is one of these exceptions as it is absent in 27 out of 130 strains (Supplementary Table S1). The majority of strains also lacked the toxin that was only present among nine strains in Clade 2.
Besides these virulence factors, a number of other proteins may be involved in the virulence of C. pseudotuberculosis and were detected in the data set. The genes encoding PknD (cp12CS0282_00131), a serine/threonine protein kinase involved in pathogenesis of M. tuberculosis [54], a hypothetical protein (cp12CS0282_00875) which is highly abundant in the C. pseudotuberculosis 12CS0282 and PknG (cp12CS0282_00897, serine/threonine protein kinase) a putative vaccine target [55] were also highly conserved within the data set.
3.2. Proteome Analyses
3.2.1. C. Pseudotuberculosis 12CS0282 Whole Cell Proteome, Surface Fraction and Secreted Proteome Fraction
The theoretical proteome of C. pseudotuberculosis strain 12CS0282 comprises 2174 unique proteins of which 70% were located to the cytoplasm, 20% are membrane associated and 10% of the proteins are predicted to be secreted. Twenty-three percent include transmembrane helices (TMH), 5% a SPI secretion signal, 3% are secreted lipoproteins including a SPII signal sequence and 1% are secreted via a TAT secretion system.
By mass spectrometry, we were able to identify 1444 proteins, representing 66.4% of the theoretical proteome. A total of 1317 proteins were detected in the whole proteome, 1083 proteins in the surface fraction obtained by trypsin treatment and 187 proteins in the secreted proteins fraction with an overlap of 159 proteins. Obviously, the tryptic shaving procedure resulted in a high number of lysed cells, as described earlier for a C. ulcerans strain [31]. Therefore, data sets were curated using localization prediction approaches and cytoplasmic proteins were eliminated from surface and secreted proteome fractions. Furthermore, only proteins identified in all three runs were considered for further analysis. In total 1023 unique proteins were found, 958 proteins from the whole cell fraction, 114 proteins from the surface fraction and 33 secreted proteins were present in all three runs and 12 overlapping proteins were found (Figure 4).
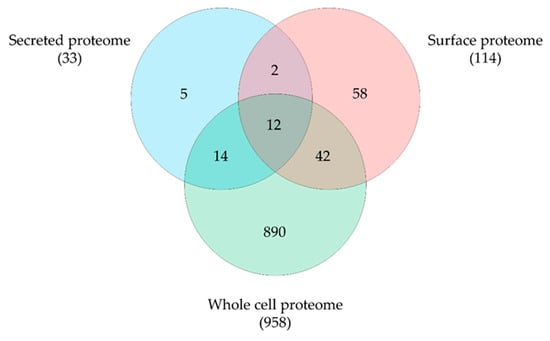
Figure 4.
C. pseudotuberculosis 12CS0282 proteins identified in this study. Distribution and overlap of proteins in the different fractions analyzed. Data sets were curated using localization prediction approaches and cytoplasmic proteins were eliminated from surface and secreted proteome fractions.
3.2.2. Metabolic Pathway Analysis
The proteins identified were classified in respect to their metabolic function (Figure 5, Table 3). When the proteins of the secretome were analyzed in detail, protein Cp12CS0282_00875 represented 69.9 ± 3.1% of the total protein content. This putative trypsin-like serine protease has a homology with Vsp2, a known virulence factor from C. ulcerans, and is highly conserved within the analyzed C. pseudotuberculosis strains (Supplementary Table S1).

Figure 5.
Metabolic pathways of identified proteins in the whole cell proteome (left), surface fraction (middle) and extracellular proteome (right). Each area displays one protein and equal to the relative abundance. Pathways are separated in metabolism (green), information storage and processing (purple), cellular processes and signaling (blue), environmental information processing (orange), genetic information processing (yellow), poorly characterized (grey), uncharacterized (black), and in pathogenicity (red).
Table 3.
Metabolic pathway analysis.
Independent from the pathway, a similar percentage of the theoretical proteome and the identified proteins was found, while proteins related to pathogenicity and poorly and uncharacterized proteins were slightly over-represented. The theoretical proteome and the overall identified protein pool included 3.1% of protein related to pathogenicity, while 3.9% of proteins found in all biological replicates were attributed to this group. Moreover, a high number of poorly characterized and uncharacterized proteins were found, summing up to 42.6% of the theoretical proteome, 45.7% of all identified proteins and 46.0% of proteins found in all replicates.
In respect to the main metabolic pathways, all proteins from the glycolysis, pentose-phosphate pathway, synthesis of the cell envelope, glycolysis, gluconeogenesis, tricarboxylic acid (TCA) cycle, heme biosynthesis, fatty acid synthesis and amino acid synthesis were found except enzymes involved in the synthesis of phenylalanine and tyrosine.
3.2.3. Identification of Virulence Proteins
The respective proteins of almost all annotated virulence genes identified in the 12CS0282 genome were identified in different proteome fractions when the extracellular secretome fraction, cell-wall-bound surface fraction and whole cell fraction were analyzed (Table 4), indicating that this strain is already preadapted to host contact and pathogenicity-related proteins do not have to be expressed upon host contact.
Table 4.
Validation and distribution of C. pseudotuberculosis 12CS0282 virulence factors. Proteins encoded by virulence genes in Table 1 were analyzed in respect to presence, localization (E, extracellular, S, surface; W, whole cell fraction) and relative abundance (% of protein content in fraction).
3.2.4. Reverse Vaccinology
From the cellular and extracellular proteome of C. pseudotuberculosis strain 12CS0282 only experimentally identified proteins from the surface fraction and secreted proteome were considered for further analyses in a reverse vaccinology approach. The theoretical proteome of C. pseudotuberculosis strain 12CS0282 was filtered for proteins located on the surface and secreted proteins. From these, only proteins from the core proteome of C. pseudotuberculosis were selected for prediction of probable vaccine targets (Vaxign2), antigenic potential (VaxiJen) and filtered for non-host homologous proteins and essential proteins. The described reverse vaccinology approach (Figure 1) revealed 22 probable vaccine and drug targets (Table 5). Four of these, Ndh (cp12CS0282_01097), SenX3 (cp12CS0282_00370), FtsI (cp12CS0282_01491) and YidC (cp12CS0282_00093) were also identified as putative vaccines targets in a former in silico analysis of C. pseudotuberculosis [45].
Table 5.
Predicted vaccine and drug targets. The table shows the protein ID of the corresponding protein from C. pseudotuberculosis strain 12CS0282, the protein name, molecular weight (MW) and predicted stability (+: stable; −: unstable).
An analysis using a protein-protein interactions network revealed promising vaccine and drug targets (Figure 6). Both potential vaccine and drug targets Pdp4 (cp12CS0282_00932) and FtsI (cp12CS0282_01491) show interactions with DacA, Fts proteins and Mur proteins. Fts and Mur proteins are involved in construction of the cell wall and for cell division (Figure 6a,b). Two probable vaccine and drug targets have a direct interaction SecD (cp12CS0282_01259) and YidC1 (cp12CS0282_00093). Both proteins are involved in bacterial secretion (Sec proteins) and cell division (Fts proteins) (Figure 6c).
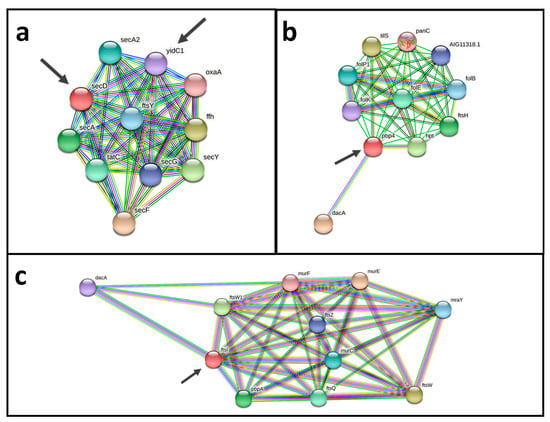
Figure 6.
Protein-protein interaction networks of putative vaccine and drug targets. (a) The two probable vaccine and drug targets SecD (cp12CS0282_01259) and YidC1 (cp12CS0282_00093), have direct interactions and are interacting with components of bacterial protein secretion (Sec proteins) and cell division (Fts proteins). (b,c) Pdp4 (cp12CS0282_00932) and FtsI (cp12CS0282_01491) show interactions with DacA, Fts proteins and Mur proteins, which are involved in cell wall synthesis and cell division. Putative vaccine and drug targets identified here are indicated by arrows.
3.2.5. Interaction with Macrophages
The ability of pathogens to invade and survive in host cells is fundamental for host colonization. In order to determine intracellular viability in THP-1 cells, gentamicin protection assays were carried out using C. glutamicum ATCC13032 as a non-pathogenic corynebacterium, C. ulcerans 809 and C. silvaticum W25 as pathogenic members of the genus together with C. pseudotuberculosis strains 12CS0282 and FRC41. THP-1 cells were infected with bacteria at MOI 1 for 30 min. Subsequently, the cells were washed and further incubated for 2, 4 and 20 h in cell culture medium containing gentamicin to kill extracellular bacteria, then detached and lysed. The ratio of colony forming units (CFU) in inoculum and lysates provided the percentage of invasive bacteria (Figure 7a). Furthermore, the ratio of internalized viable bacteria (after 2 h infection) and CFU from lysates from 4 h and 20 h infections gave the percentage of time-dependent survival within the phagocytes (Figure 7b).
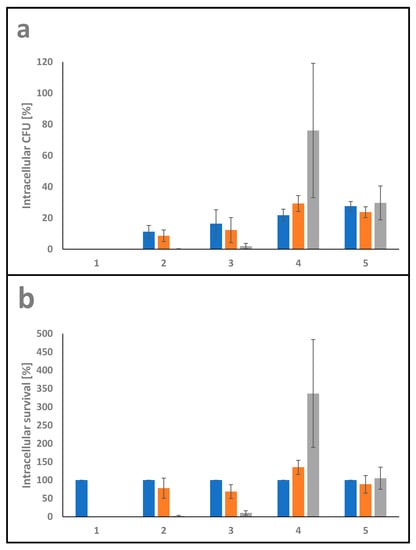
Figure 7.
Quantitative analysis of C. pseudotuberculosis–macrophage interaction. THP-1 cells were infected with C. ulcerans 809 (2), C. silvaticum W25 (3), C. pseudotuberculosis 12CS0282 (4) and C. pseudotuberculosis FRC41 (5) at MOI 1 for 30 min. Infection with the non-pathogenic C. glutamicum ATCC13032 (1) served as a negative control. In order to kill extracellular bacteria, cells were incubated with medium containing gentamicin and after 2 (blue), 4 (orange) and 20 h (grey), cells were harvested, lysed and lysates were plated on blood agar plates to recover intracellular CFU. (a) Intracellular CFU in percent referred to the inoculum. (b) Intracellular survival in percent referred to the bacteria that were taken up after 2 h.
The intracellular survival rate of the non-pathogenic C. glutamicum ATCC13032 was 0% after 2 h incubation, indicating that C. glutamicum was not able to resist phagolysis by THP-1 cells and is quickly eradicated. C. ulcerans 809 had the lowest uptake rate with 11 ± 4% followed by C. silvaticum W25 with 16 ± 9% on average. The C. pseudotuberculosis strains reached values between 22 ± 4% and 28 ± 3%.
The CFU of C. ulcerans and C. silvaticum gradually decreased with a survival rate of about 78 ± 28% and 69 ± 19% after 4 h incubation and only 2 ± 2% and 10 ± 6% after 20 h incubation, respectively. While C. pseudotuberculosis strain FRC41, isolated from a 12 years old girl with necrotizing lymphadenitis, did not multiply in the macrophages, strain 12CS0282 showed significant growth inside the human phagocyte cells used with survival rates of 135 ± 19% after 4 h incubation and 337 ± 147% on average after 20 h of incubation.
4. Discussion
Although C. pseudotuberculosis is a member of the toxigenic corynebacteria [2], sequences of tox gene-encoding corynephages are rarely detectable in the available genomes. Less than 10% of the strains are tox gene carriers. Nevertheless, the species is an important animal pathogen and obviously also a potential danger for human health.
The C. pseudotuberculosis strain 12CS0282 investigated here showed a higher resistance against human phagocytes than pathogenic C. ulcerans [56,57], C. pseudotuberculosis FRC41 [9] and C. silvaticum [58] strains. Strain 12CS0282 was not only able to survive for more than 20 h within human macrophages, but could even multiply within the phagocytes. Either the strain is able to survive the harsh conditions within the macrophage, or it is able to inhibit phagolysosome maturation as shown for C. diphtheriae and C. ulcerans [59,60]. A prime candidate for macrophage damage is the sphingomyelinase phospholipase D (PLD), which was shown to promote survival of C. pseudotuberculosis in a murine macrophage cell line [61]. Another possible candidate is the trypsin-like serine protease Cp12CS0282_00875, which is the by far most abundant secreted protein. However, although intriguing, it is most likely not a single virulence factor, but a combination of proteins and liposaccharides may determine the pathogenic potential of the bacteria, since virulence-related processes such as adhesion, invasion and intracellular survival seem to be multifactorial mechanisms in corynebacteria [62].
As demonstrated by proteome analyses, almost all known virulence factors for C. pseudotuberculosis were expressed already under standard laboratory conditions and without any host contact, indicating a high degree of pre-adaptation to host conditions putatively also enabling fast response to phagocytosis and supporting survival within the macrophages. As an approach to get further information on proteins important for host cell interaction and survival in macrophages proteome analyses of bacteria internalized by phagocytes may be carried out in future studies.
In the light of the pathogenic potential of the species, vaccination may be the gold standard to inhibit infection and colonization of host species and different targets were already published [13,14,15]. To contribute to this field, we used a reverse vaccinology approach similar to a study of Araújo and co-workers [45] and were able to validate four the previously identified targets [45] also for strain 12CS0282. The reverse vaccinology approach carried out here hint to especially three promising putative vaccine targets, FtsI, SecD and YidC. FtsI is a transpeptidase required for synthesis of peptidoglycan, involved in cell cycle control and cell division of growing bacteria and predicted to be a potential drug target in M. tuberculosis [63]. SecD is involved in protein secretion in M. tuberculosis [64] and contributes to the virulence potential in Listeria monocytogenes [65] and Staphylococcus aureus [66]. YidC is an essential insertase protein, which mediates the assembly and insertion of inner membrane proteins. The function is related to folding and insertion of lipoproteins in the plasma membrane [67] and controls respiratory metabolism in M. tuberculosis [68]. In addition, the identified proteins may serve as excellent drug targets, since they interfere with cell envelope synthesis and protein secretion. As our dataset was filtered for essential proteins, it is a shortcoming of this study that these targets cannot be easily verified by gene deletion experiments. Either methods for gene knock-downs have to be developed for this organism, or proteins have to be overexpressed, purified and used for antibody production to test their suitability in animal studies, as descried earlier for C. pseudotuberculosis proteins Cp09 and Cp40 [14].
As an alternative to the described proteome and reverse vaccinology approach, immunopeptidomics may help to develop vaccines directed against C. pseudotuberculosis. Immunopeptidomics approaches focus on antigen discovery by the detection of peptides that are presented at the surface of cells by major histocompatibility complexes. In principle, this allows an untargeted identification of bacterial antigens. However, up to now, only a few-mainly intracellular-bacterial pathogens have been investigated by this approach. The reason for this is the main drawback of the technique, the large number of infected cells required for proteome analysis [69].
In summary, our study provides a further example of the strength of combining different -omics and bioinformatics approaches as demonstrated earlier [45] and may help to develop strategies to combat this important animal pathogen, which also has a pathogenic potential against humans.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/proteomes10040039/s1, Figure S1: Phylogenetic tree of C. pseudotuberculosis. Table S1: Distribution of virulence genes in C. pseudotuberculosis. Table S2: Mass spectrometry data of 12CS0208 proteins.
Author Contributions
Conceptualization, J.M. and A.B.; methodology, J.H.; investigation, J.M. and M.B.; resources, J.H.; data curation, J.M. and V.S.; writing—original draft preparation, V.S, J.M. and A.B.; writing—review and editing, A.B.; visualization, J.M.; supervision, A.B.; project administration, A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The genome sequences of strains C. pseudotuberculosis 12CS0282 has been submitted to GenBank (Accession no.: NZ_CP074370.1). Raw data files and msf files of mass spectrometric analyses were deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [70]. Data are available via ProteomeXchange PXD036354 (project webpage: http://www.ebi.ac.uk/pride/archive/projects/PXD036354; FTP download: ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2022/11/PXD036354; published 21 November 2022).
Acknowledgments
Isolate 12CS0282 was kindly provided by Helmut Hotzel (Friedrich-Löffler-Institut, Jena, Germany).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sangal, V.; Hoskisson, P.A. Corynephages: Infections of the infectors. In Diphtheria and its Etiological Agents; Burkovski, A., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 67–82. [Google Scholar]
- Riegel, P.; Ruimy, R.; Brie, D.; De Prkost, G.; Jehl, F.; Christen, R. Taxonomy of Corynebacterium diphtheriae and related taxa with recognition of Corynebacterium ulcerans sp. nov. nom. rev. FEMS Microbiol. Lett. 1995, 126, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Dorella, F.A.; Pacheco, L.G.C.; Oliveira, S.C.; Miyoshi, A.; Azevedo, V. Corynebacterium pseudotuberculosis: Microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet. Res. 2006, 37, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Baird, G.J.; Fontaine, M.C. Corynebacterium pseudotuberculosis and its role in ovine caseous lymphadenitis. J. Comp. Pathol. 2007, 137, 179–201. [Google Scholar] [PubMed]
- Silva, A.; Schneider, M.P.; Cerdeira, L.; Barbosa, M.S.; Ramos, R.T.; Carneiro, A.R.; Santos, R.; Lima, M.; D’Afonseca, V.; Almeida, S.S.; et al. Complete genome sequence of Corynebacterium pseudotuberculosis I19, a strain isolated from a cow in Israel with bovine mastitis. J. Bacteriol. 2011, 193, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Windsor, P.A.; Bush, R.D. Caseous lymphadenitis: Present and near forgotten from persistent. Small Ruminant Res. 2016, 142, 6–10. [Google Scholar] [CrossRef]
- Selim, S.A. Oedematous skin disease of buffalo in Egypt. J. Vet. Med. B Infect. Dis. Vet. Public Health. 2001, 48, 241–258. [Google Scholar] [CrossRef]
- Peel, M.M.; Palmer, G.G.; Stacpoole, A.M.; Kerr, T.G. Human lymphadenitis due to Corynebacterium pseudotuberculosis: Report of ten cases from Australia and review. Clin. Infect. Dis. 1997, 24, 185–191. [Google Scholar] [CrossRef]
- Trost, E.; Ott, L.; Schneider, J.; Schröder, J.; Jaenicke, S.; Goesmann, A.; Husemann, P.; Stoye, J.; Dorella, F.A.; Rocha, F.S.; et al. The complete genome sequence of Corynebacterium pseudotuberculosis FRC41 isolated from a 12-year-old girl with necrotizing lymphadenitis reveals insights into gene- regulatory networks contributing to virulence. BMC Genom. 2010, 11, 728. [Google Scholar] [CrossRef]
- Pacheco, L.G.; Slade, S.E.; Seyffert, N.; Santos, A.R.; Castro, T.L.; Silva, W.M.; Santos, A.V.; Santos, S.G.; Farias, L.M.; Carvalho, M.A.; et al. A combined approach for comparative exoproteome analysis of Corynebacterium pseudotuberculosis. BMC Microbiol. 2011, 11, 12. [Google Scholar] [CrossRef]
- Silva, W.M.; Seyffert, N.; Ciprandi, A.; Santos, A.V.; Castro, T.L.; Pacheco, L.G.; Barh, D.; Le Loir, Y.; Pimenta, A.M.; Miyoshi, A.; et al. Differential exoproteome analysis of two Corynebacterium pseudotuberculosis biovar ovis strains isolated from goat (1002) and sheep (C231). Curr. Microbiol. 2013, 67, 460–465. [Google Scholar] [CrossRef]
- Soares, S.C.; Silva, A.; Trost, E.; Blom, J.; Ramos, R.; Carneiro, A.; Ali, A.; Santos, A.R.; Pinto, A.C.; Diniz, C.; et al. The Pan-Genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS ONE 2013, 8, e53818. [Google Scholar]
- Silva, W.M.; Carvalho, R.D.; Soares, S.C.; Bastos, I.F.; Folador, E.L.; Souza, G.H.; Le Loir, Y.; Miyoshi, A.; Silva, A.; Azevedo, V. Label-free proteomic analysis to confirm the predicted proteome of Corynebacterium pseudotuberculosis under nitrosative stress mediated by nitric oxide. BMC Genom. 2014, 15, 1065. [Google Scholar]
- Silva, J.W.; Droppa-Almeida, D.; Borsuk, S.; Azevedo, V.; Portela, R.W.; Miyoshi, A.; Rocha, F.S.; Dorella, F.A.; Vivas, W.L.; Padilha, F.F.; et al. Corynebacterium pseudotuberculosis Cp09 mutant and Cp40 recombinant protein partially protect mice against caseous lymphadenitis. BMC Vet. Res. 2014, 10, 965. [Google Scholar] [CrossRef] [PubMed]
- Droppa-Almeida, D.; da Silva, G.A.; Gaspar, L.M.D.A.C.; Pereyra, B.B.S.; Nascimento, R.J.M.; Borsuk, S.; Franceschi, E.; Padilha, F.F. Peptide vaccines designed with the aid of immunoinformatic against Caseous Lymphadenitis promotes humoral and cellular response induction in mice. PLoS ONE 2021, 16, e0256864. [Google Scholar]
- Parise, D.; Teixeira Dornelles Parise, M.; Pinto Gomide, A.C.; Figueira Aburjaile, F.; Bentes Kato, R.; Salgado-Albarrán, M.; Tauch, A.; Ariston de Carvalho Azevedo, V.; Baumbach, J. The Transcriptional Regulatory Network of Corynebacterium pseudotuberculosis. Microorganisms 2021, 9, 415. [Google Scholar] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lio, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Goker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Tange, O. GNU Parallel—The command-line power tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Möller, J.; Schorlemmer, S.; Hofmann, J.; Burkovski, A. Cellular and extracellular proteome of the animal pathogen Corynebacterium silvaticum, a close relative of zoonotic Corynebacterium ulcerans and Corynebacterium pseudotuberculosis. Proteomes 2020, 8, 19. [Google Scholar] [CrossRef]
- Kraner, M.E.; Müller, C.; Sonnewald, U. Comparative proteomic profiling of the choline transporter-like1 (CHER1) mutant provides insights into plasmodesmata composition of fully developed Arabidopsis thaliana leaves. Plant J. 2017, 92, 696–709. [Google Scholar]
- Bittel, M.; Gastiger, S.; Amin, B.; Hofmann, J.; Burkovski, A. Surface and extracellular proteome of the emerging pathogen Corynebacterium ulcerans. Proteomes 2018, 6, 18. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Meth. 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.; Kraner, M.; Sonnewald, U.; Sangal, V.; Tittlbach, H.; Winkler, J.; Winkler, T.H.; Melnikov, V.; Lang, R.; Sing, A.; et al. Proteomics of diphtheria toxoid vaccines reveals multiple proteins that are immunogenic and may contribute to protection of humans against Corynebacterium diphtheriae. Vaccine 2019, 37, 3061–3070. [Google Scholar] [PubMed]
- Tjalsma, H.; Lambooy, L.; Hermans, P.W.; Swinkels, D.W. Shedding & shaving: Disclosure of proteomic expressions on a bacterial face. Proteomics 2008, 8, 1415–1428. [Google Scholar]
- Schäfer, W.; Eckart, R.A.; Schmid, B.; Cagköylü, H.; Hof, K.; Muller, Y.A.; Amin, B.; Lührmann, A. Nuclear trafficking of the anti-apoptotic Coxiella burnetii effector protein AnkG requires binding to p32 and Importin-α 1. Cell. Microbiol. 2017, 19, e12634. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.R.; Melli, G.; Sahinalp, S.C.; Yu, N.Y.; Lo, R.; Dao, P.; Brinkman, F.S.L.; Wagner, J.R.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Nielsen, H.; Brunak, S.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef]
- Bernhardt, J.; Funke, S.; Hecker, M.; Siebourg, J. Visualizing Gene Expression Data via Voronoi Treemaps. In Proceedings of the Sixth International Symposium on Voronoi Diagrams, Copenhagen, Denmark, 23–26 June 2009; pp. 233–241. [Google Scholar] [CrossRef]
- Otto, A.; Bernhardt, J.; Meyer, H.; Schaffer, M.; Herbst, F.-A.; Siebourg, J.; Mäder, U.; Lalk, M.; Hecker, M.; Becher, D. Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nature Commun. 2010, 1, 137. [Google Scholar] [CrossRef] [PubMed]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual account of protein investment in cellular functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- Araújo, C.L.; Alves, J.; Nogueira, W.; Pereira, L.C.; Gomide, A.C.; Ramos, R.; Azevedo, V.; Silva, A.; Folador, A. Prediction of new vaccine targets in the core genome of Corynebacterium pseudotuberculosis through omics approaches and reverse vaccinology. Gene 2019, 702, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Chukwu-Osazuwa, J.; Cao, T.; Vasquez, I.; Gnanagobal, H.; Hossain, A.; Machimbirike, V.I.; Santander, J. Comparative Reverse Vaccinology of Piscirickettsia salmonis, Aeromonas salmonicida, Yersinia ruckeri, Vibrio anguillarum and Moritella viscosa, Frequent Pathogens of Atlantic Salmon and Lumpfish Aquaculture. Vaccines 2022, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.T.; Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef]
- Barh, D.; Jain, N.; Tiwari, S.; Parida, B.P.; D’Afonseca, V.; Li, L.; Ali, A.; Santos, A.R.; Guimarães, L.C.; de Castro Soares, S.; et al. A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chem. Biol. Drug Des. 2011, 78, 73–84. [Google Scholar] [CrossRef]
- Ong, E.; Cooke, M.F.; Huffman, A.; Xiang, Z.; Wong, M.U.; Wang, H.; Seetharaman, M.; Valdez, N.; He, Y. Vaxign2: The second generation of the first Web-based vaccine design program using reverse vaccinology and machine learning. Nucleic Acids Res. 2021, 49, W671–W678. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. Predicting class I major histocompatibility complex (MHC) binders using multivariate statistics: Comparison of discriminant analysis and multiple linear regression. J. Chem. Inf. Model. 2007, 47, 234–238. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Goker, M.; Sproer, C.; Klenk, H.P. When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 2013, 195, 413–418. [Google Scholar] [PubMed]
- Be, N.A.; Bishai, W.R.; Jain, S.K. Role of Mycobacterium tuberculosis PknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiol. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Thais de Oliveira Silva, M.; Barros de Pinho, R.; da Rocha Fonseca, B.; Silvestre Brilhante Bezerra, F.; Severo Sabedra Sousa, F.; Kommling Seixas, F.; Collares, T.; Meyer Nascimento, J.R.; Wagner Portela, R.; Ariston Carvalho Azevedo, V.; et al. NanH and PknG putative virulence factors as a recombinant subunit immunogen against Corynebacterium pseudotuberculosis infection in mice. Vaccine 2020, 38, 8099–8106. [Google Scholar] [PubMed]
- Trost, E.; Al-Dilaimi, A.; Papavasiliou, P.; Schneider, J.; Viehoever, P.; Burkovski, A.; de Castro Soares, S.; Silva Almeida, S.; Alves Dorella, F.; Miyoshi, A.; et al. Comparative analysis of two complete Corynebacterium ulcerans genomes and detection of candidate virulence factors. BMC Genom. 2011, 12, 383. [Google Scholar]
- Hacker, E.; Ott, L.; Schulze-Luehrmann, J.; Lührmann, A.; Wiesmann, V.; Wittenberg, T.; Burkovski, A. The killing of macrophages by Corynebacterium ulcerans. Virulence 2016, 7, 45–55. [Google Scholar]
- Möller, J.; Musella, L.; Melnikov, V.; Geißdörfer, W.; Burkovski, A.; Sangal, V. Phylogenomic characterisation of a novel corynebacterial species pathogenic to animals. Antonie Van Leeuwenhoek 2020, 113, 1225–1239. [Google Scholar]
- Ott, L.; Hacker, E.; Kunert, T.; Karrington, I.; Etschel, P.; Lang, R.; Wiesmann, V.; Wittenberg, T.; Singh, A.; Varela, C.; et al. Analysis of Corynebacterium diphtheriae macrophage interaction: Dispensability of corynomycolic acids for inhibition of phagolysosome maturation and identification of a new gene involved in synthesis of the corynomycolic acid layer. PLoS ONE 2017, 12, e0180105. [Google Scholar]
- Weerasekera, D.; Hahn, J.; Herrmann, M.; Burkovski, A. Induction of necrosis in human macrophage cell lines by Corynebacterium diphtheriae and Corynebacterium ulcerans strains isolated from fatal cases of systemic infections. Int. J. Mol. Sci. 2019, 20, 4109. [Google Scholar] [CrossRef]
- McKean, S.C.; Davies, J.K.; Moore, R.J. Expression of phospholipase D, the major virulence factor of Corynebacterium pseudotuberculosis, is regulated by multiple environmental factors and plays a role in macrophage death. Microbiology 2007, 153, 2203–2211. [Google Scholar]
- Tauch, A.; Burkovski, A. Molecular armory or niche factors: Virulence determinants of Corynebacterium species. FEMS Microbiol. Lett. 2015, 362, fnv185. [Google Scholar] [CrossRef]
- Slayden, R.A.; Belisle, J.T. Morphological features and signature gene response elicited by inactivation of FtsI in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2009, 63, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.K.; Zulauf, K.E.; Braunstein, B. The Sec Pathways and Exportomes of Mycobacterium tuberculosis. Microbiol. Spec. 2017, 2, 24. [Google Scholar]
- Burg-Golani, T.; Pozniak, Y.; Rabinovich, L.; Sigal, N.; Nir Paz, R.; Herskovits, A.A. Membrane chaperone SecDF plays a role in the secretion of Listeria monocytogenes major virulence factors. J. Bacteriol. 2013, 195, 5262–5272. [Google Scholar] [PubMed]
- Quiblier, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Berger-Bächi, B.; Senn, M.M. Contribution of SecDF to Staphylococcus aureus resistance and expression of virulence factors. BMC Microbiol. 2011, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Petriman, N.A.; Jauß, B.; Hufnagel, A.; Franz, L.; Sachelaru, I.; Drepper, F.; Warscheid, B.; Koch, H.G. The interaction network of the YidC insertase with the SecYEG translocon, SRP and the SRP receptor FtsY. Sci. Rep. 2018, 8, 578. [Google Scholar]
- Thakur, P.; Choudhary, E.; Pareek, M.; AgarWal, N. Regulation and overexpression studies of YidC in Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 17114. [Google Scholar]
- Mayer, R.L.; Impens, F. Immunopeptidomics for next-generation bacterial vaccine development. Trends Microbiol. 2021, 29, 1034–1045. [Google Scholar]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, 442–450. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).