1. Introduction
In clinical treatment of neuronal medicine, various types of neuronal cells are reconstructed or transplanted into patients who suffer severe neuronal diseases or injuries such as Amyotrophic Lateral Sclerosis (ALS) [
1], Parkinson’s disease [
2,
3] and spinal cord injury [
4,
5,
6,
7,
8]. In the case of spinal cord injury, for example, reconstruction of the damaged neuronal cells colony might be carried out first if the damage is not so severe. On the other hand, in the future, regenerative medicine using pluripotency of stem cells will be a probable way to determine whether the damage severe to such an extent that the spinal cord is totally snapped off. Through the cellular reprogramming technique, by using four transcription factors Oct3/4, Sox2, Klf4, c-Myc, induced pluripotent stem (iPS) cells have been developed [
9,
10,
11] that possess inherent properties to proliferate infinitely while retaining the ability to differentiate into almost any type of cell. Regenerative medicine is in currently in a phase of rapid development and the engraftment of transplanted neuronal cells, as well as the reconstruction of the neuronal network, will become the most important issue. In both cases it is strongly needed to develop new approaches or device systems to support these medical treatments technically, as well as to promote efficiency and quickness of the treatments.
Several studies have reported that well-controlled dynamic stimulations are able to promote abilities of cell proliferation, growth and recovery from damage [
12,
13,
14,
15,
16,
17]. Likewise, the appropriate mechanical stimulations may have the potential to raise the efficiency and speed of the differentiation of the iPS cells into the objective cells. Limited research reports have been published so far about the relationship between mechanical stimulations and cellular differentiation of rat pheochromocytoma (PC12), embryonic stem (ES) cells or iPS cells [
18,
19,
20,
21,
22,
23,
24]. Rapid progresses in microelectromechanical systems (MEMS) to manipulate minute living cells, and in advanced materials to be used for scaffold in three-dimensional cellular culturing provide a novel method to approach various sciences and technologies related to iPS cells [
25,
26]. Effects of low frequency (10–40 Hz), low magnitude vibrations in the differentiation of stem cells have recently been reported, stating that low frequency, low magnitude vibrations are sufficient to induce or to promote mesenchymal stem cells into neural cells [
27,
28,
29].
In view of the circumstances, in this study, a novel in vitro simulator in which micro dynamic stimulations are applied to an artificially damaged neuronal cell colony is proposed in order to investigate the possibility of promoting the reconstruction of a damaged neuronal cell colony. Firstly, a cantilever-type piezoelectric mini-vibration table, which enables us to impose various micro dynamic stimulations directly onto the cultured adhesive cells or cellular colonies on culture dish set inside the incubator, is developed together with an in vitro seeding-damage-reconstruction device. Secondly, in order to verify the ability of the developed system, dynamic stimulations are applied to a damaged mouse neuronal cell colony. The neuronal cells differentiated from mouse iPS cells are cultured for 5 days, and after the cells have grown sufficiently, the neuronal cell colony with neurites are physically damaged by cutting off treatment. Micro dynamic stimulations by using the developed mini-vibration table are applied for 6 days after the cutting damage, while NeuroFluor NeuO(STEMCELL Technologies) is used to conduct fluorescent staining of the living neuronal cells. Thirdly, effects of the dynamic stimulations on the reconstructing process of the damaged neuronal cells are investigated by analysing the immunostained areas of the living neuronal cell colony with or without dynamic stimulations. The parameters of reconstruction efficiency as well as the extensibility of reconstructed neurites are newly proposed in order to achieve quantitative analysis about the possibility of promoting the reconstruction of a damaged neuronal cell colony.
3. Results
Effects of the dynamic stimulations on reconstructing process of the damaged neuronal cell colony differentiated from iPS cells are estimated by measuring the immunostained areas of the neural cells with or without the micro dynamic stimulations by utilizing the developed vibration table. In the results shown in the following Figures 7 and 10, differences between the dynamically stimulated case and the static case (control, i.e., at 0 Hz) are statistically analysed by t-test and are considered to be significant when the probability, p value, is less than 5 percent.
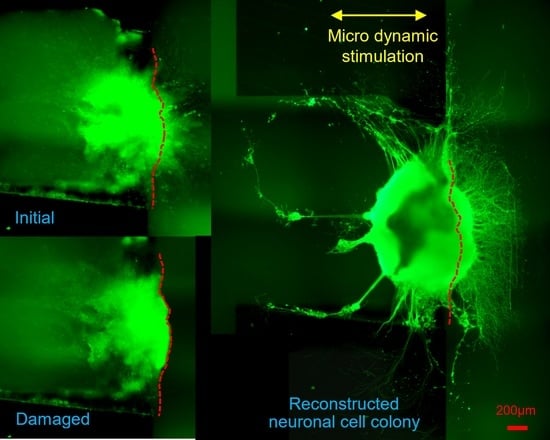
Figure 6 shows immunostained images of the in vitro reconstructing process of the damaged neuronal cell colony. Left side figures are the static case (control, without micro dynamic stimulation) for comparison. Right side figures are the case under micro dynamic stimulation (vibration total amplitude: 60 µm, frequency: 5 Hz, 12 h. interval). The reason why frequency of 5 Hz is used in this experiment is that it is most effective in differentiation and growth of mouse iPS cells into neurons as found by our previous study [
23].
Figure 6a–d are stained fluorescent image by NeuroFluor NeuO, which has a remarkable ability to stain living neurons without fixation, and that the fluorescent image is continually observed under phase-contrast microscope throughout the experiments. As for
Figure 6e, on the other hand, βⅢ-tubulin is stained after fixation of the cell colony in order to obtain a stable and clear image. It is clearly confirmed that neurites are sprouted around the seeding pit. Figures in green show direct fluorescent microscopic images with a dotted red line, which shows the approximate cutting line given from (b). The right end side figures in red show binarized images of the lost area (Alost in (a)) or the reconstructed area (Areconst. in (c), (d), (e)) in case of micro dynamic stimulation by utilizing WinROOF (scale bars = 200 µm). Furthermore, the yellow arrow in
Figure 6c–e shows the direction of vibration stimulation by utilizing the developed mini-vibration table.
Figure 6a shows the image just before cutting damage was made at day 5.
Figure 6b shows the state immediately after the cutting damage (day 5), while the dotted red line shows the approximate cutting line. Black shadowy line behind the red cutting line shows the right-side edge of the Plate A explained in
Figure 4.
Figure 6c shows the initial reconstructing state (day 7 or 2 days after cutting damage) Neuronal reconstruction around the damaged boundary is gradually taking place by sprouting neurites in several points.
Figure 6d shows the middle of reconstructing process (day 9 or 4 days after cutting damage) where reconstructed neurites are growing almost evenly and densely.
Figure 6e shows the reconstructed final image in this series of experiment (day 11 or 6 days after cutting damage) where neurites are dramatically grown in numbers, density and length, especially in direction to the right-hand along with the cutting edge. This seems prominent in case under the micro dynamic stimulation.
Figure 7 shows a comparison of reconstruction efficiency Areconst./Alost (computed area ratio from
Figure 6) of the damaged neuronal cell colony between the statically cultured neurons and the dynamically stimulated ones (vibration total amplitude: 60 µm, frequency: 5 Hz, 12 h. interval) with respect to culture time after the cutting damage operation. The reconstruction efficiency increases with increasing culture time as 7, 9 and 11 days (or, 2, 4 and 6 days after cutting damage) in the stimulation cases, while the situation is not observed in the static cases. In all three cases, the reconstruction efficiency of the dynamically stimulated cases is prominently higher than that of the static ones. Significant differences are observed between the statically cultured damaged neurons and the dynamically stimulated ones as the culture time increases, such as 9 to 11 days (or 4 to 6 days after the cutting damage). At day 11 (6 days after cutting damage), it is found that the reconstructed area from the cutting damage is up to around 80% in the dynamically stimulated case, while the area is almost unchanged with culture days and shows around 25% in the static case.
Figure 8 shows the relationship between the increase rate of reconstruction efficiency Areconst./Alost against the static case and the culture time.
Y-axis is normalized reconstruction efficiency (stimulation/static) calculated from the results shown in
Figure 7. The increase rate of reconstruction efficiency increases exponentially as the culture time increases, such as 7, 9, 11 days (or, 2, 4 and 6 days after cutting damage). The increase rate is the highest in the day 11 with about 241%, while it is about 96% in the day 7. It is found that the more the culture time after the cutting damage increases, the more the increase rate of reconstruction efficiency increases dramatically. This suggests that not only imposing the dynamic stimulation but also having appropriate culture time should be key factors to promote the reconstruction of damaged neuronal cell colony.
Figure 9 shows definition of parameters for evaluation of extensibility of reconstructed neurites of the colony [=(E
m/Elost all) × 100] based on the typical binarized immunostained images which is treated by utilizing image analyser software WinROOf, Image J and Octave.
Figure 9a shows definition of Llost_max: maximum straight-line length of neuron colony measured from edge line of the cutting damage to the furthest front line of the lost neurites, while E
lost all: number of all pixels of the lost area of neurons Alost.
Figure 9b shows parameters L
m and E
m defined on the reconstructed neurons colony. The parameter L
m = (m/100) × Llost_max shows the location line until which the neuronal pixels are counted for from the line of cutting damage, with m = 100 coincide with Llost_max. The parameter E
m is the number of neuronal pixels existing on the right side of line L
m on reconstructed neurites of the colony.
Figure 10 shows the effect of dynamic stimulation (vibration amplitude: 60 µm, frequency: 5 Hz, 12 h. interval) upon extensibility of the reconstructed neurites [=(E
m/Elost all) × 100] with respect to the value of m (indicating location line up to which the neuronal pixels are counted for) as well as the culture time. Results are computed based on the parameters defined in
Figure 9. At day 5 (
Figure 10a), extensibilities of the statically cultured damaged neurons and the dynamically stimulated ones almost coincide with each other, since these are the initial state, i.e., just before the cutting damage. In
Figure 10b–d, significant differences are observed between the statically cultured damaged neurons and the dynamically stimulated ones as the culture time increases, such as day 7, 9, 11, respectively (or 2, 4, 6 days after cutting damage). Especially, at day 11 (or 6 days after cutting damage), it is found that significant differences are observed in the cases with lower m such as m = 15~35, which correspond to comparatively proximal area to the cutting damage line. Furthermore, at day 11, maximum value of m in the stimulated cases reaches up to 110, which implies that reconstructed neurites finally surpass the initial length: Llost_max. (straight-line length measuring from the line of cutting damage to the front line). Thus, it is confirmed that the dynamic stimulation promotes reconstruction and extension of neurites within the damaged neuronal cell colony.
Figure 11 shows the effect of culture time after cutting damage upon the extensibility of reconstructed neurites [=(E
m/Elost all) × 100] in the static case (a) and the dynamically stimulated (vibration amplitude: 60 µm, frequency: 5 Hz, 12 h. interval) case (b). Basically, figure axis and each data are the same as those given in
Figure 10. On day 7, the extensibility of the reconstructed neurites in the dynamically stimulated case (
Figure 11b) is far below compared to the initial state which is the state immediately before the cutting damage. However, the extensibility gradually increases as the culture time increases in order of 7, 9, 11 days (or 2, 4, 6 days after cutting damage), and reaches closer to the initial state. On the other hand, in the static case (
Figure 11a), the extensibility does not increase systematically or stably as the culture time increases in order of 7, 9, 11, and it is not clear whether the extensibility is closer to the initial state or not, as the culture time increases. Especially, on the day 11, in the lower area of m, such as 0 to 22 which is the proximal area to the cutting damage, the extensibility is far below compared to the case of day 9.
4. Discussion
It has been pointed out that neuronal cells are equipped with the function of a sensor that senses physical or chemical stimulations and elongates neurites in appropriate directions to connect nearby neuronal cells in forming a network [
37], while the cells are generally said to be hard to recover from major physical damage where the cell body or main neurites are almost severed completely. However, in the case of neuronal cell colony differentiated from iPS cells with appropriate size and environment [
7,
8], neurites might newly sprout from near-by undamaged neuronal cells within the colony. By avoiding the damaged neurons or their remains, some of the newly sprouted neurites will have the ability to make a connection between the damaged host neurons and the target neurons, similar to rewiring activity taking place in the spinal cord injury [
4,
5]. The immunostained images of the in vitro reconstructing process of the damaged neuronal cell colony shown in
Figure 6c–e of this study imply the above possibility. Especially,
Figure 6e shows how the reconstructing activity takes place around the damaged edge area of the neuronal cell colony. It is notable that an abundant thick bundle of neurites are sprouted from the right-hand side periphery of the cell colony and these construct a complex and dense neurites network. On the other hand, these phenomena are not seen in other un-damaged edge areas of the colony.
In spinal cord injury, it has been pointed out that three cases are likely to try to rewire the partly injured spine [
5]. The first case is to promote axonal regeneration or direct endogenous reconnection, and the second one is to have axonal sprouting or indirect endogenous reconnection. The third case is to conduct neural stem cell transplantation and that to promote indirect exogenous reconnection, which will be valid even in the fully damaged spine. In the first two approaches, activation of damaged axon as well as neuron itself will likely be key factor for regenerating the damaged axons and following successful endogenous reconnection. Therefore, it is indispensable to develop techniques and devices to promote axonal regeneration and axonal sprouting.
Regenerative medicine, on the other hand, is currently in a phase of rapid development where the engraftment of transplanted neuronal cells as well as the reconstruction of neuron networks are becoming the most important issues to be solved. In both cases, henceforth, it is strongly needed to develop new approaches or device systems to support these medical treatments technically, as well as to promote efficiency and quickness of the treatments. In practical cases, various electric massagers embedded in bags, pillows, sofas, beds, etc. in which electric vibrators are utilized might become simple and possible candidates to apply the dynamic stimulations to the patient who has been under the clinical regenerative treatments, provided that appropriate technical adjustments and medical monitoring are carefully conducted.
In
Figure 7, it is found that the reconstruction efficiency increases with increasing culture time, such as in 7, 9 and 11 days (or, 2, 4 and 6 days after cutting damage) in the stimulation cases. Furthermore, the increase rate of the reconstruction efficiency also increases with elapsed culture time as shown in
Figure 8. On the other hand, in the static cases, the reconstruction efficiency is almost unchanged with increasing culture time as 7, 9 and 11 days. This implies that an important fact for the neuronal cell colony reconstruction from the cutting damage such that the appropriately controlled dynamic stimulation is indispensable to initiate the reconstruction process. However, the biochemical and molecular mechanism of the reconstruction phenomena of the damaged neuronal cells has not been elucidated thoroughly [
4,
5,
6,
7,
8]. In this study, the neurons used are directly differentiated from iPS cells and that undamaged neurons near-by damaged area in the neuronal cell colony might intrinsically retain better ability in neurites sprouting and growth compared to spontaneously grown neuronal cell colony.
In
Figure 10 and
Figure 11, it is observed that the extensibility of the reconstructed neurites in the dynamically stimulated case is gradually increases as the culture time increases in the order of 7, 9, 11 days (or 2, 4, 6 days after cutting damage), and is getting closer to the initial state (the state immediately before the cutting damage), while the phenomenon does not explicitly occur in the static case. This means that the appropriately controlled dynamic stimulation is quite effective and stable to promote the extensibility of the reconstructed neurites from the damage. It is considered that the sensors of neuronal cells detect the vibration stimulation and translate it into chemical or electrical signals, and these might be transferred to neuronal cell growth cones leading stable neurites sprouting.
The results obtained in this study suggest various opportunities to develop effective reconstruction strategies of neurites from the physical damage. Namely, it is confirmed that the appropriate dynamic stimulation (or in other words dynamic stress) even as µm order amplitude vibration (acceleration is computed as 3.0 × 10−3 G from Equation (6)) is a promising approach to promote reconstruction of the damaged neuronal cell colony. The method can possibly provide a supporting measure in clinical treatment for the damaged neurons in cases such as spinal cord injury, in which a huge scientific and medical challenge to restore lost functions is still towering.
In the future, a more realistic and comprehensive in vitro simulator should be developed to meet serious and urgent clinical needs, for instance, in spinal cord injuries. In order to achieve this goal, firstly, it is necessary to identify the optimal condition of the directionality, the frequency, the magnitude, the timing, and the duration time of the applied dynamic stimulations to promote the reconstruction of the damaged neuronal cell colony. Secondly, a human neuronal cell colony should be used instead of the mouse one, although the differentiation time from human iPS cells into neuronal cells is required 30 to 40 days and its process is much more complicated. Thirdly, the present in vitro simulator system should be expanded in a three-dimensional culture environment in order to realize more actual conditions of the spinal cord injury. One promising candidate for this challenge might be the incorporation of gel-embedded three-dimensional culture [
24]. Finally, neuroelectric reactions between the reconstructed neurons and nearby target neurons should be examined to confirm not only structural reconstruction but also functional reconstruction as a neuronal network.
5. Conclusions
In this study, a novel in vitro simulator which is able to apply micro dynamic stimulations onto a damaged neuronal cell colony differentiated from iPS cells is developed in order to investigate neuronal repair and regeneration. By using the system, as a first step, influence of the dynamic stimulations on the promotion of the reconstruction process of the neuronal cell colony from the damage is investigated. Newly proposed parameters for the reconstruction efficiency as well as the extensibility of reconstructed neurites are used for the evaluation of the present method. The results can be summarized as follows.
(1) Applying dynamic stimulations by utilizing a developed mini-vibration table together with an in vitro seeding-damage-reconstruction device and time dependent fluorescent staining observations by using NeuroFluor NeuO are able to provide a novel method to simulate ongoing reconstruction process of the damaged neuronal cell colony;
(2) It is found that significant differences are observed in the reconstruction efficiency: Areconst./Alost between the statically cultured damaged neuronal cell colony and the dynamically stimulated one as the culture time increasing such as 4 to 6 days after cutting damage operation;
(3) The extensibility of the reconstructed neurites [=(Em/Elost all)×100] in the dynamically stimulated case is gradually increasing as the culture time increasing in the order of 2, 4, 6 days after cutting damage operation and is getting closer to the initial state, while it does not explicitly occur in the static case;
(4) It is confirmed that applying appropriate micro dynamic stimulations is a promising approach to promote the reconstruction of the damaged neuronal cell colony, and that the present in vitro simulator system could possibly provide some fundamental information as a simple disease model for the spinal cord injury.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












