Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs

Abstract
:1. Introduction
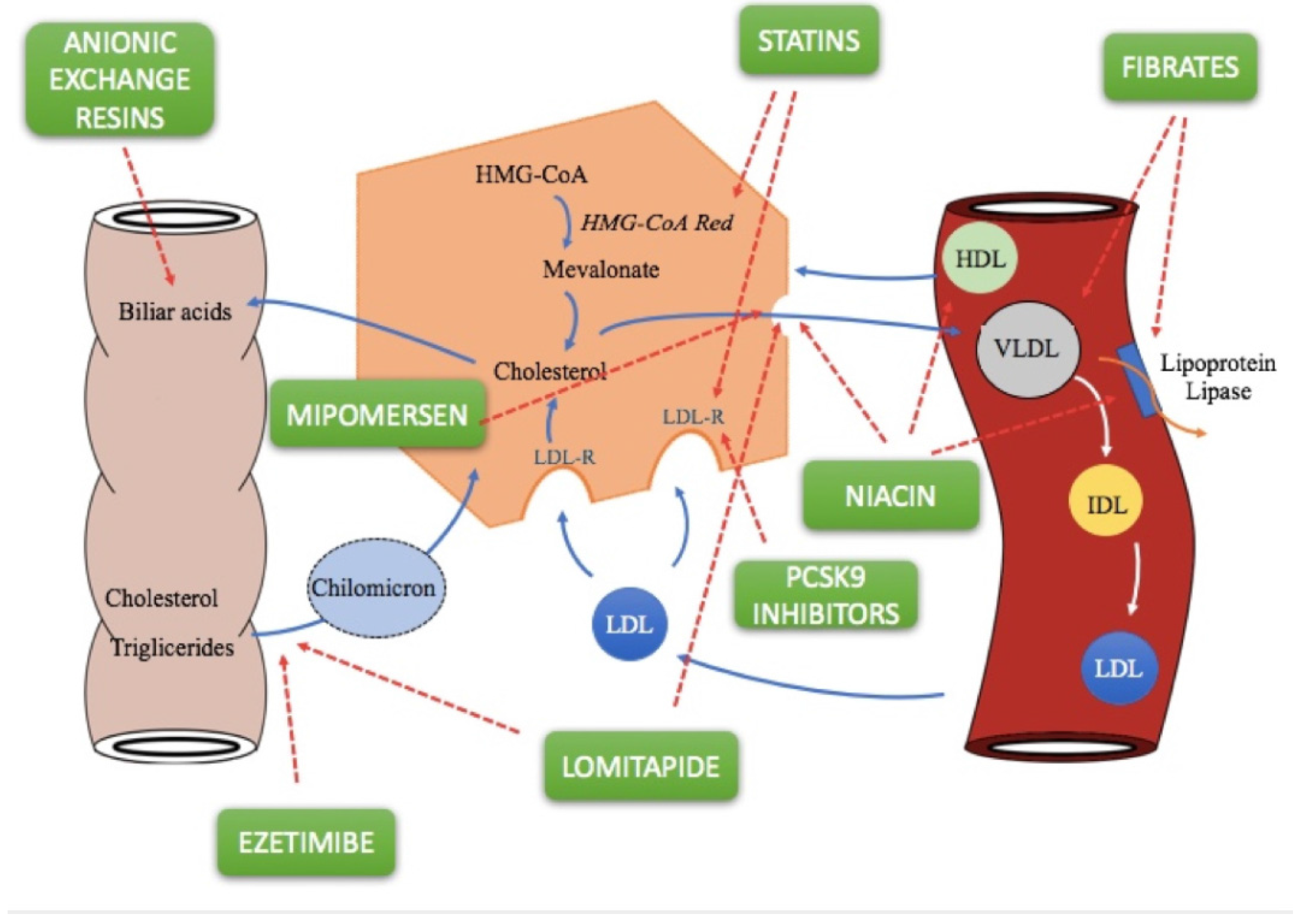
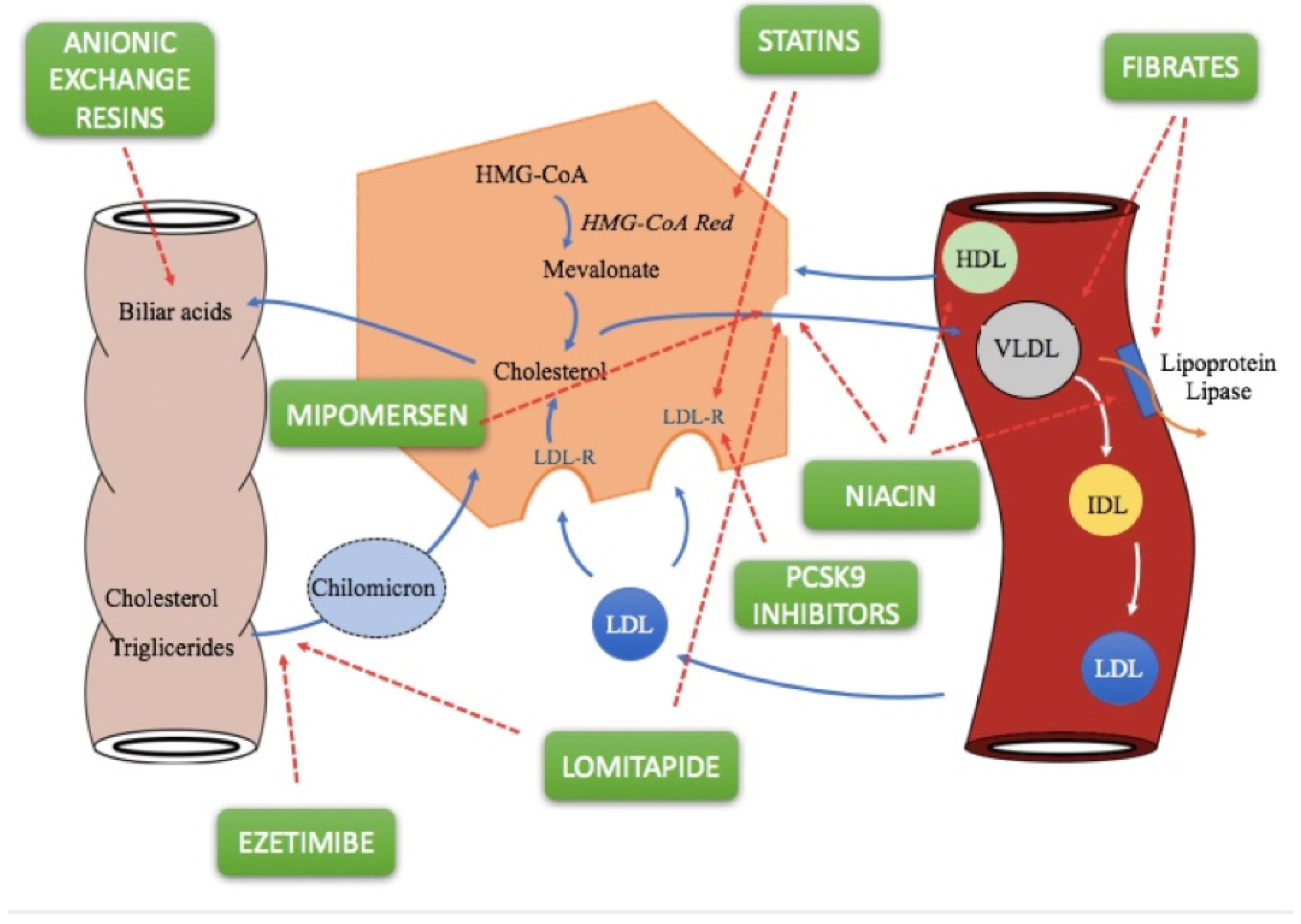
2. Lipid-Lowering Drugs
2.1. Statins
2.2. Fibrates
2.3. Bile Acid Sequestrants
2.4. Ezetimibe
2.5. Niacin
2.6. Omega-3 Fatty Acids
2.7. PCSK9 Inhibitors
2.7.1. Alirocumab
2.7.2. Evolocumab
2.8. Lomitapide
2.9. Mipomersen
3. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Iaea, D.B.; Maxfield, F.R. Cholesterol trafficking and distribution. Essays Biochem. 2015, 57, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.P. Subclinical atherosclerosis: What it is, what it means and what we can do about it. Int. J. Clin. Pract. 2008, 62, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Treatment of high blood cholesterol in, executive summary of the third report of the national cholesterol Education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- European Association for Cardiovascular. ESC/EAS Guidelines for the management of dyslipidaemias: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur. Heart J. 2011, 32, 1769–1818. [Google Scholar]
- Kulik, A.; Brookhart, M.A.; Levin, R.; Ruel, M.; Solomon, D.H.; Choudhry, N.K. Impact of statin use on outcomes after coronary artery bypass graft surgery. Circulation 2008, 118, 1785–1792. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [PubMed]
- Raghow, R. Statins redux: A re-assessment of how statins lower plasma cholesterol. World J. Diabetes 2017, 8, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Stender, S.; Schuster, H.; Barter, P.; Watkins, C.; Kallend, D.; MERCURY I Study Group. Comparison of rosuvastatin with atorvastatin, simvastatin and pravastatin in achieving cholesterol goals and improving plasma lipids in hypercholesterolaemic patients with or without the metabolic syndrome in the MERCURY I trial. Diabetes Obes. Metab. 2005, 7, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Khokhar, M. Mechanism of action of anti-hypercholesterolemia drugs and their resistance. Eur. J. Pharmacol. 2014, 741, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Gazzerro, P.; Proto, M.C.; Gangemi, G.; Malfitano, A.M.; Ciaglia, E.; Pisanti, S.; Santoro, A.; Laezza, C.; Bifulco, M. Pharmacological actions of statins: A critical appraisal in the management of cancer. Pharmacol. Rev. 2012, 64, 102–146. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.D.; Raal, F.J.; Stein, E.A.; Rader, D.J.; Blasetto, J.; Palmer, M.; Wilpshaar, W. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis 2008, 197, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, D.; Li, D.B.; Yu, X.; Shi, G.B. Comparison of the efficacy and safety of intensive-dose and standard-dose statin treatment for stroke prevention: A meta-analysis. Medicine (Baltimore) 2016, 95, e4950. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Evans, M.A. Statin adverse effects: A review of the literature and evidence for a mitochondrial mechanism. Am. J. Cardiovasc. Drugs 2008, 8, 373–418. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Feely, J. Pharmacokinetic-pharmacodynamic drug interactions with HMG-CoA reductase inhibitors. Clin. Pharmacokinet. 2002, 41, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A. Plasma lipoproteins: Genetic influences and clinical implications. Nat. Rev. Genet. 2009, 10, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Ewang-Emukowhate, M.; Wierzbicki, A.S. Lipid-lowering agents. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Schulz, I. Treatment of dyslipidemia: How and when to combine lipid lowering drugs. Arq. Bras. Endocrinol. Metabol. 2006, 50, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Armitage, J.; Bowman, L. Lipid-lowering treatment: Today’s recommended management. Prescriber 2006, 17, 33–44. [Google Scholar] [CrossRef]
- Ling, H.; Luoma, J.T.; Hilleman, D. A Review of Currently Available Fenofibrate and Fenofibric Acid Formulations. Cardiol. Res. 2013, 4, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.B.; Spence, J.D. Clinical pharmacokinetics of fibric acid derivatives (fibrates). Clin. Pharmacokinet. 1998, 34, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Moutzouri, E.; Kei, A.; Elisaf, M.S.; Milionis, H.J. Management of dyslipidemias with fibrates, alone and in combination with statins: Role of delayed-release fenofibric acid. Vasc. Health Risk Manag. 2010, 6, 525–539. [Google Scholar] [PubMed]
- Bezafibrate Infarction Prevention Study. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000, 102, 21–27. [Google Scholar]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z.; Adler, Y.; Shemesh, J.; Tanne, D.; Leor, J.; Boyko, V.; Schwammenthal, E.; Behar, S. Effect of bezafibrate on incidence of type 2 diabetes mellitus in obese patients. Eur. Heart J. 2005, 26, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z.; Tanne, D.; Boyko, V.; Behar, S. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch. Intern. Med. 2005, 165, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, J.; Yamamoto, K.; Ageta, M. Effects of fenofibrate on high-density lipoprotein particle size in patients with hyperlipidemia: A randomized, double-blind, placebo-controlled, multicenter, crossover study. Clin. Ther. 2002, 24, 1614–1626. [Google Scholar] [CrossRef]
- Kornitzer, M.; Dramaix, M.; Vandenbroek, M.D.; Everaert, L.; Gerlinger, C. Efficacy and tolerance of 200 mg micronised fenofibrate administered over a 6-month period in hyperlipidaemic patients: An open Belgian multicenter study. Atherosclerosis 1994, 110, S49–S54. [Google Scholar] [CrossRef]
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-González, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, CD009753. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, D.R.; Bacon, S. Influence of lovastatin plus gemfibrozil on plasma lipids and lipoproteins in patients with heterozygous familial hypercholesterolemia. Circulation 1989, 79, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Staffa, J.A.; Chang, J.; Green, L. Cerivastatin and reports of fatal rhabdomyolysis. N. Engl. J. Med. 2002, 346, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.H.; Davidson, M.H. Reporting rate of rhabdomyolysis with fenofibrate + statin versus gemfibrozil + any statin. Am. J. Cardiol. 2005, 95, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Robidoux, J. Dyslipidemia management update. Curr. Opin. Pharmacol. 2017, 33, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, K.L.; Schonder, K.S. Colesevelam: Potential uses for the newest bile resin. Cardiovasc. Drug. Rev. 2005, 23, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Pizzoferrato, M.; Ponziani, F.R.; Gasbarrini, G.; Gasbarrini, A. Use and indications of cholestyramine and bile acid sequestrants. Intern. Emerg. Med. 2013, 8, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H. A systematic review of bile acid sequestrant therapy in children with familial hypercholesterolemia. J. Clin. Lipidol. 2011, 5, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Armani, A.; Toth, P.P. Colesevelam hydrochloride in the management of dyslipidemia. Expert Rev. Cardiovasc. Ther. 2006, 4, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H. The use of colesevelam hydrochloride in the treatment of dyslipidemia: A review. Expert Opin. Pharmacother. 2007, 8, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Bove, M.; Fogacci, F.; Cicero, A.F.G. Pharmacokinetic drug evaluation of ezetimibe + simvastatin for the treatment of hypercholesterolemia. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Lipka, L.J. Ezetimibe: A first-in-class, novel cholesterol absorption inhibitor. Cardiovasc. Drug Rev. 2003, 21, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H.; McGarry, T.; Bettis, R.; Melani, L.; Lipka, L.J.; LeBeaut, A.P.; Suresh, R.; Sun, S.; Veltri, E.P. Ezetimibe coadministered with simvastatin in patients with primary hypercholesterolemia. J. Am. Coll. Cardiol. 2002, 40, 2125–2134. [Google Scholar] [CrossRef]
- Gille, A.; odor, E.T.; Ahmed, K.; Offermanns, S. Nicotinic acid: Pharmacological effects and mechanisms of action. Ann. Rev. Pharmacol. Toxicol. 2008, 48, 79–106. [Google Scholar] [CrossRef] [PubMed]
- Bodor, E.T.; Offermanns, S. Nicotinic acid: An old drug with a promising future. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S68–S75. [Google Scholar] [CrossRef] [PubMed]
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [PubMed]
- Ho, C.K.; Walker, S.W. Statins and their interactions with other lipid-modifying medications: Safety issues in the elderly. Ther. Adv. Drug Saf. 2012, 3, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.S.; Miller, M.; Tighe, A.P.; Davidson, M.H.; Schaefer, E.J. Omega-3 fatty acids and coronary heart disease risk: Clinical and mechanistic perspectives. Atherosclerosis 2008, 197, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.M.; Myung, S.K.; Lee, Y.J.; Seo, H.G.; Korean Meta-analysis Study Group. Efficacy of omega-3 fatty acid supplements (eicosapentaenoic acid and docosahexaenoic acid) in the secondary prevention of cardiovascular disease: A meta-analysis of randomized, double-blind, placebo-controlled trials. Arch. Intern. Med. 2012, 172, 686–694. [Google Scholar] [PubMed]
- Bays, H.E.; Ballantyne, C.M.; Kastelein, J.J.; Isaacsohn, J.L.; Braeckman, R.A.; Soni, P.N. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the Multi-center, plAcebo-controlled, Randomized, double-blINd, 12-week study with an open-label Extension [MARINE] trial). Am. J. Cardiol. 2011, 108, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Fujisue, K.; Tsujita, K. Current status of lipid management in acute coronary syndrome. J. Cardiol. 2017, 70, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M. PCSK9 inhibition in the management of familial hypercholesterolemia. J. Cardiol. 2017, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Descamps, O.S.; Fraass, U.; Dent, R.; März, W.; Gouni-Berthold, I. Anti-PCSK9 antibodies for hypercholesterolaemia: Overview of clinical data and implications for primary care. Int. J. Clin. Pract. 2017, 71. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Garg, J.; Shah, N.; Sumner, A. PCSK9 inhibitors: A new era of lipid lowering therapy. World J. Cardiol. 2017, 9, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Agency, E.E.M. Assessment report Praluent International Non-Proprietary Name: Alirocumab Procedure No. EMEA/H/C/003882/0000. EMA/CHMP/392430/2015 Committee for Medicinal Products for Human Use (CHMP), 23 July 2015. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003882/WC500194524.pdf (accessed on 5 November 2017).
- Wong, N.D.; Rosenblit, P.D.; Greenfield, R.S. Advances in dyslipidemia management for prevention of atherosclerosis: PCSK9 monoclonal antibody therapy and beyond. Cardiovasc. Diagn. Ther. 2017, 7 (Suppl. 1), S11–S20. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Pedersen, T.R.; Keech, A.C.; Sever, P.S.; Park, J.G.; Sabatine, M.S. FOURIER Steering Committee & Investigators. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: A prespecified secondary analysis of the FOURIER trial. Lancet 2017, 390, 1962–1971. [Google Scholar] [PubMed]
- Agency, E.E.M. Assessment report Repatha International Non-Proprietary name: Evolocumab. Procedure No. EMEA/H/C/003766/0000. EMA/CHMP/222019/2015. Committee for Medicinal Products for Human Use (CHMP), 21 May 2015. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/003766/WC500191400.pdf (accessed on 5 November 2017).
- Polychronopoulos, G.; Tziomalos, K. Novel treatment options for the management of heterozygous familial hypercholesterolemia. Expert Rev. Clin. Pharmacol. 2017, 10, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Kastelein, J.J. Lomitapide and mipomersen: Two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 2014, 129, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Meagher, E.A.; du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef]
- Tuteja, S.; Duffy, D.; Dunbar, R.L.; Movva, R.; Gadi, R.; Bloedon, L.T.; Cuchel, M. Pharmacokinetic interactions of the microsomal triglyceride transfer protein inhibitor, lomitapide, with drugs commonly used in the management of hypercholesterolemia. Pharmacotherapy 2014, 34, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.A.; Miyares, M.A. Lomitapide: A novel agent for the treatment of homozygous familial hypercholesterolemia. Am. J. Health Syst. Pharm. 2014, 71, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Geary, R.S. Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br. J. Clin. Pharmacol. 2013, 76, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Peer-Rev. J. Formul. Manag. 2014, 39, 119–122. [Google Scholar]
- Ricotta, D.N.; Frishman, W. Mipomersen: A safe and effective antisense therapy adjunct to statins in patients with hypercholesterolemia. Cardiol. Rev. 2012, 20, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Sjouke, B.; Balak, D.M.; Beuers, U.; Ratziu, V.; Stroes, E.S. Is mipomersen ready for clinical implementation? A transatlantic dilemma. Curr. Opin. Lipidol. 2013, 24, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Fulcher, J.; O’Connell, R.; Voysey, M.; Emberson, J.; Blackwell, L.; Mihaylova, B.; Simes, J.; Collins, R.; Kirby, A.; et al. Efficacy and safety of LDL-lowering therapy among men and women: Meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar] [PubMed]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Goff, D.C., Jr.; Lloyd-Jones, D.M.; Smith, S.C., Jr.; Blum, C.; Schwartz, J.S.; 2013 ACC/AHA Cholesterol Guideline Panel. Treatment of blood cholesterol to reduce atherosclerotic cardiovascular disease risk in adults: Synopsis of the 2013 American College of Cardiology/American Heart Association cholesterol guideline. Ann. Intern. Med. 2014, 160, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Langslet, G.; Emery, M.; Wasserman, S.M. Evolocumab (AMG 145) for primary hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2015, 13, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Bessac, L.; Berdan, L.G.; Bhatt, D.L.; Bittner, V.; Diaz, R.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: Rationale and design of the ODYSSEY outcomes trial. Am. Heart J. 2014, 168, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A.; Maki, K.C.; Orringer, C.E.; Jones, P.H.; Kris-Etherton, P.; Sikand, G.; La Forge, R.; Daniels, S.R.; Wilson, D.P.; Morris, P.B.; et al. National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia: Part 2. J. Clin. Lipidol. 2015, 9 (Suppl. 6), S1–S122.e1. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Z.; De Bacquer, D.; Kotseva, K.; Prugger, C.; De Backer, G.; Wood, D.; EUROASPIRE III Study Group. Treatment potential for dyslipidaemia management in patients with coronary heart disease across Europe: Findings from the EUROASPIRE III survey. Atherosclerosis 2013, 231, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.V.; Brown, A.S.; Ference, B.A.; Toth, P.P.; Underberg, J.A. Genetic effects on efficacy of lipid-lowering drugs. J. Clin. Lipidol. 2017, 11, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Toth, P.P.; Worthy, G.; Gandra, S.R.; Sattar, N.; Bray, S.; Cheng, L.I.; Bridges, I.; Worth, G.M.; Dent, R.; Forbes, C.A.; et al. Systematic Review and Network Meta-Analysis on the Efficacy of Evolocumab and Other Therapies for the Management of Lipid Levels in Hyperlipidemia. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Cornier, M.A.; Eckel, R.H. Non-traditional dosing of statins in statin-intolerant patients-is it worth a try? Curr. Atheroscler. Rep. 2015, 17, 475. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Khan, H.; Heydon, E.; Shroufi, A.; Fahimi, S.; Moore, C.; Stricker, B.; Mendis, S.; Hofman, A.; Mant, J.; et al. Adherence to cardiovascular therapy: A meta-analysis of prevalence and clinical consequences. Eur. Heart J. 2013, 34, 2940–2948. [Google Scholar] [CrossRef] [PubMed]
- Kazi, D.S.; Moran, A.E.; Coxson, P.G.; Penko, J.; Ollendorf, D.A.; Pearson, S.D.; Tice, J.A.; Guzman, D.; Bibbins-Domingo, K. Cost-effectiveness of PCSK9 inhibitor therapy in patients with heterozygous familial hypercholesterolemia or atherosclerotic cardiovascular disease. JAMA 2016, 316, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.L.; Sisson, E.M.; Butler, M.; Higbea, A.; Muoio, B.; Turner, B. Lomitapide and mipomersen: Novel lipid-lowering agents for the management of familial hypercholesterolemia. J. Cardiovasc. Nurs. 2014, 29, E7–E12. [Google Scholar] [CrossRef] [PubMed]
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zodda, D.; Giammona, R.; Schifilliti, S. Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs. Pharmacy 2018, 6, 10. https://doi.org/10.3390/pharmacy6010010
Zodda D, Giammona R, Schifilliti S. Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs. Pharmacy. 2018; 6(1):10. https://doi.org/10.3390/pharmacy6010010
Chicago/Turabian StyleZodda, Donatella, Rosario Giammona, and Silvia Schifilliti. 2018. "Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs" Pharmacy 6, no. 1: 10. https://doi.org/10.3390/pharmacy6010010
APA StyleZodda, D., Giammona, R., & Schifilliti, S. (2018). Treatment Strategy for Dyslipidemia in Cardiovascular Disease Prevention: Focus on Old and New Drugs. Pharmacy, 6(1), 10. https://doi.org/10.3390/pharmacy6010010