Comprehensive Transcriptomic Analysis for Developing Seeds of a Synthetic Brassica Hexaploid


Abstract
:1. Introduction
2. Results
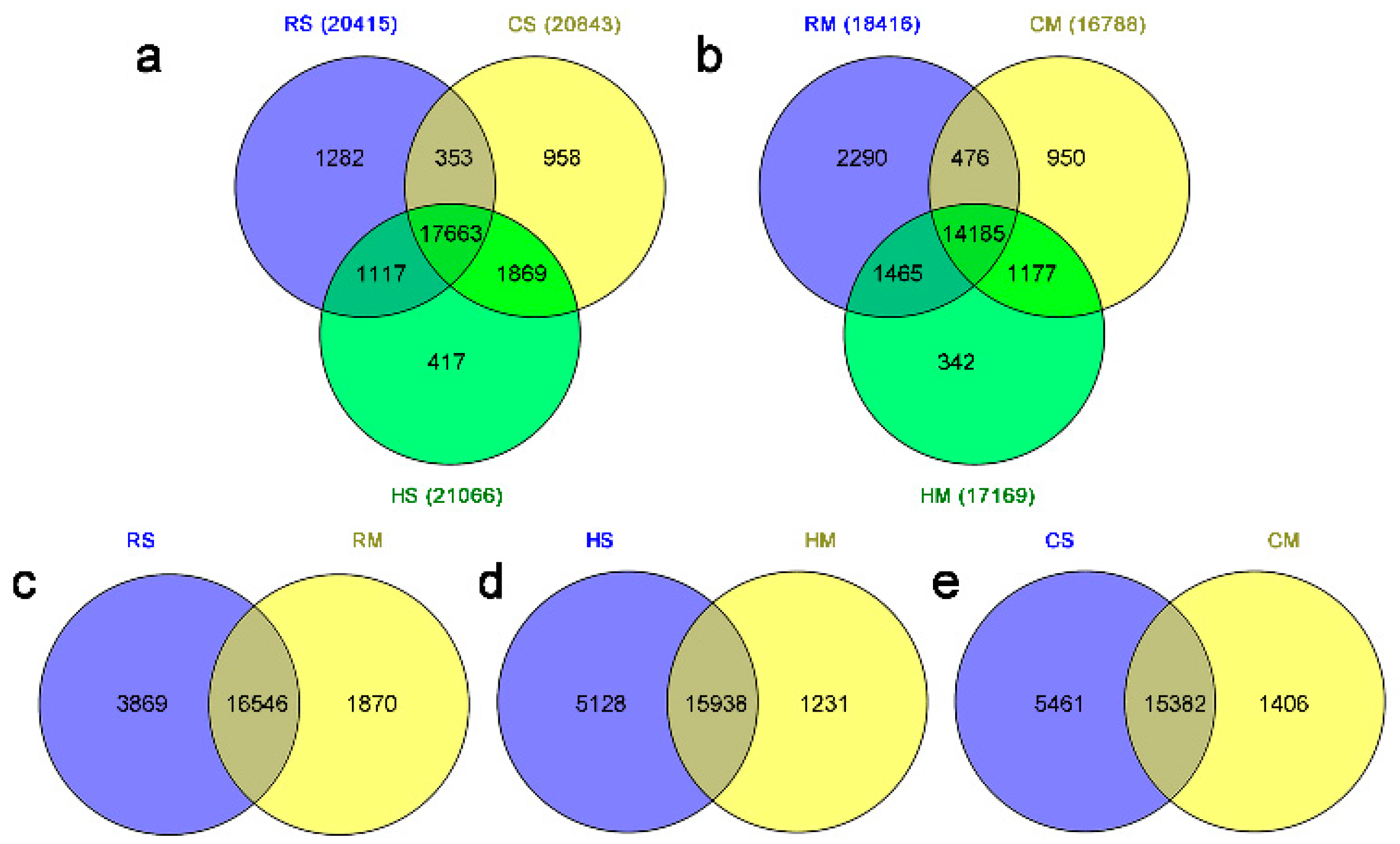
2.1. Overview of Gene Expression in the Seeds of a Brassica Hexaploid and Its Parents
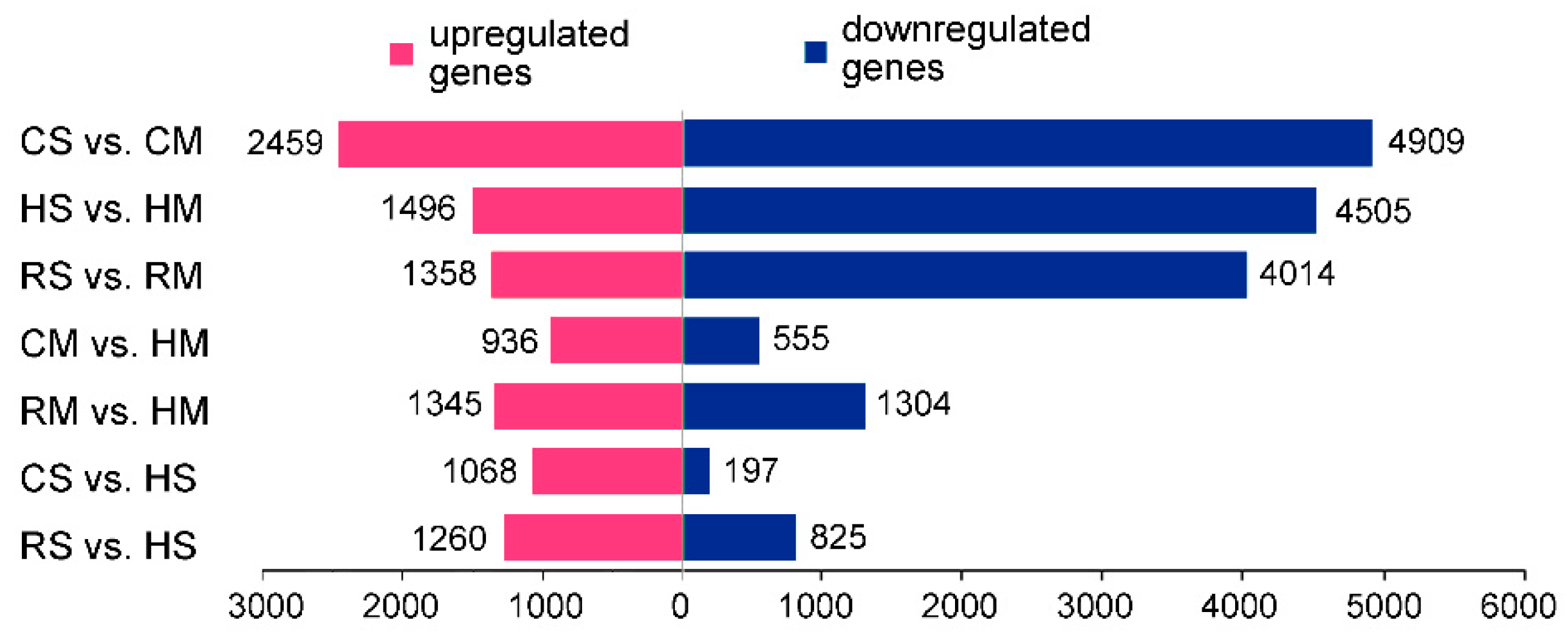
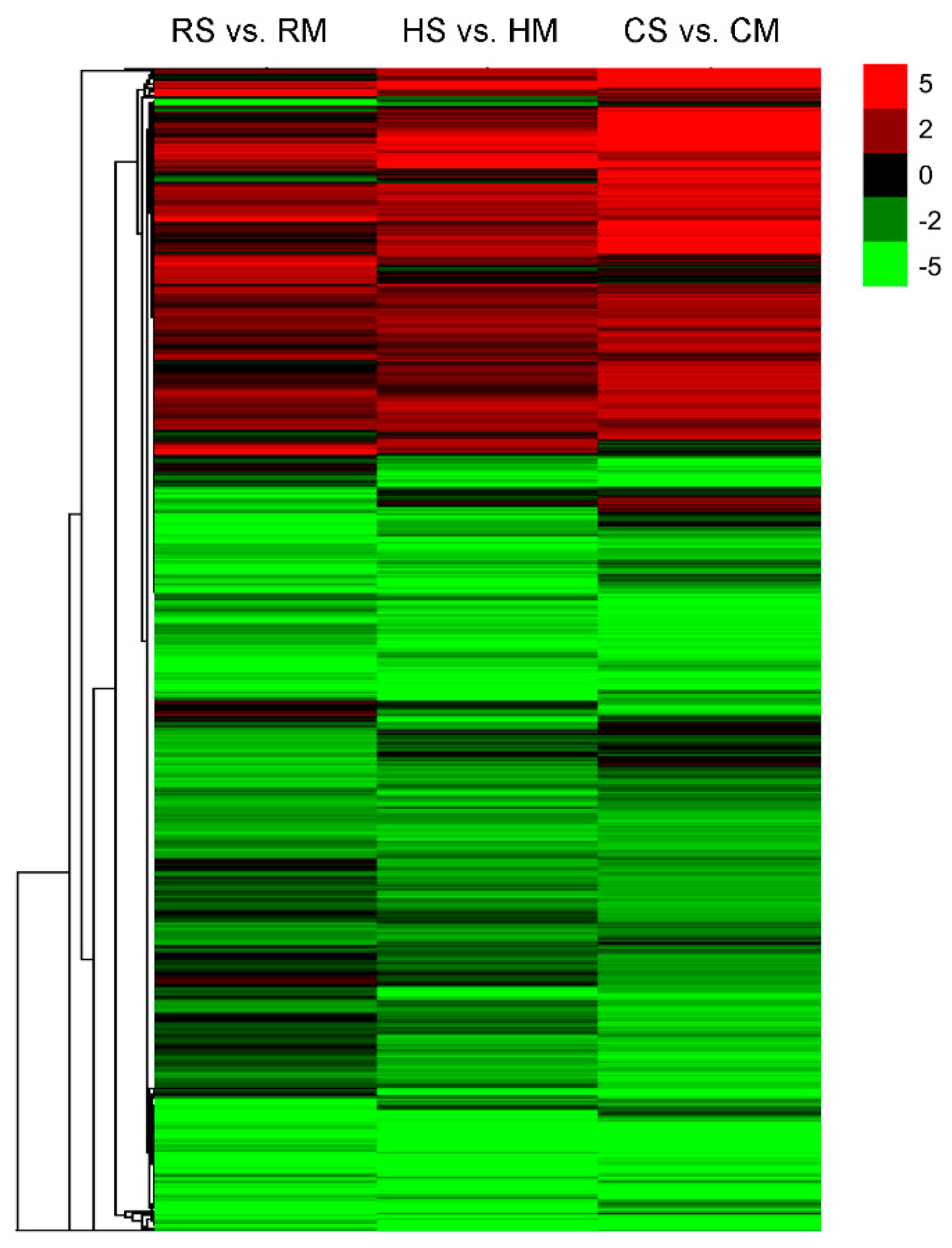
2.2. Identification of the Differentially Expressed Genes (DEGs) of Seeds among the Brassica Hexaploid and Its Parents at Two Developing Stages
2.3. Functional Annotations of the DEGs of the Seeds between the Brassica Hexaploid and Its Parents at Two Stages
2.4. Analysis of Transcription Factor (TF) Gene Expression in Seeds of the Brassica Hexaploid and Its Parents
2.5. Non-Additive Expressed Genes in Brassica Hexaploid Seeds of Two Stages
3. Discussion
3.1. DEGs May Play an Important Role in Seed Development and Maturation in a Brassica Hexaploid
3.2. Putative Transcriptome Factor Genes May Promote Better Allohexaploid Adaptation
3.3. Differential Expression of Some Genes Associated with the Synthesis of Substances Such as Flavonoids May Affect the Seed Coat Color of Mature Seeds
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, cDNA Library Construction and Illumina Sequencing
4.3. Normalized Expression Levels of Genes and Gene Annotation
4.4. Analysis of Differentially Expressed Genes (DEGs)
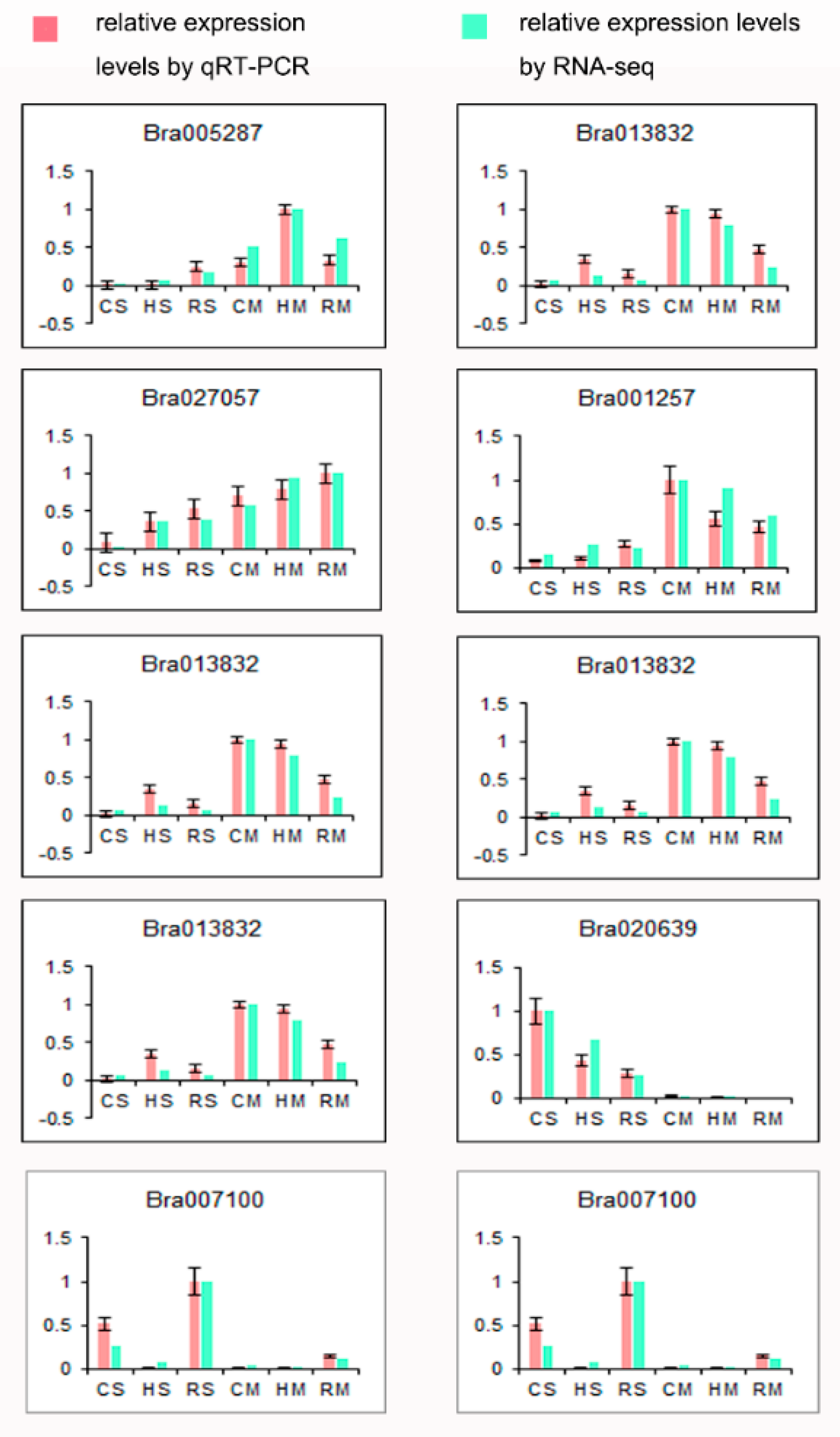
4.5. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leitch, A.R.; Leitch, I.J. Perspective—Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; McKain, M.R.; Bird, K.A.; VanBuren, R. Subgenome assignment in allopolyploids: Challenges and future directions. Curr. Opin. Plant Biol. 2018, 42, 76–80. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Han, B.; Jiao, Y. Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms. Mol. Plant 2020, 13, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 2010, 15, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Levin, D.A. Plant speciation in the age of climate change. Ann. Bot. 2019, 124, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Hahn, M.A.; van Kleunen, M.; Müller-Schärer, H. Increased phenotypic plasticity to climate may have boosted the invasion success of polyploid Centaurea stoebe. PLoS ONE 2012, 7, e50284. [Google Scholar] [CrossRef]
- Dong, Y.T.; Hu, G.J.; Yu, J.W.; Thu, S.W.; Grover, C.E.; Zhu, S.J.; Wendel, J.F. Salt-tolerance diversity in diploid and polyploid cotton (Gossypium) species. Plant J. 2020, 101, 1135–1151. [Google Scholar] [CrossRef]
- Li, N.; Xu, C.M.; Zhang, A.; Lv, R.L.; Meng, X.C.; Lin, X.Y.; Gong, L.; Wendel, J.F.; Liu, B. DNA methylation repatterning accompanying hybridization, whole genome doubling and homoeolog exchange in nascent segmental rice allotetraploids. New Phytol. 2019, 223, 979–992. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Lisch, D.; Li, Q. Creating order from chaos: Epigenome dynamics in plants with complex genomes. Plant Cell 2016, 28, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Liu, D. mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell 2014, 26, 1878–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbash, D.A.; Siino, D.F.; Tarone, A.M.; Roote, J. A rapidly evolving MYB-related protein causes species isolation in Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 5302–5307. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zou, J.; Meng, J.; Mei, S.; Wang, J. Tracing the transcriptomic changes in synthetic trigenomic allohexaploids of Brassica using an RNA-Seq approach. PLoS ONE 2013, 8, e68883. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tian, L.; Lee, H.S.; Wei, N.E.; Jiang, H.M.; Watson, B.; Madlung, A.; Osborn, T.C.; Doerge, R.W.; Comai, L.; et al. Genome wide non-additive gene regulation in Arabidopsis allotetraploids. Genetics 2006, 172, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Borpatragohain, P.; Rose, T.J.; King, G.J. Fire and brimstone: Molecular interactions between sulfur and glucosinolate biosynthesis in model and crop Brassicaceae. Front. Plant Sci. 2016, 7, 1735. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.S.; Jin, M.; Chun, J.H.; Kim, S.J.; Park, B.S.; Shon, S.H.; Kim, J.S. Functional analysis of three BrMYB28 transcription factors controlling the biosynthesis of glucosinolates in Brassica rapa. Plant Mol. Biol. 2016, 90, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Bento, M.; Pereira, H.S.; Rocheta, M.; Gustafson, P.; Viegas, W.; Silva, M. Polyploidization as a retraction force in plant genome evolution: Sequence rearrangements in Triticale. PLoS ONE 2008, 3, e1402. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, R.T.; Yoo, S.Y.; Pires, J.C.; Doerge, R.W.; Chen, Z.J.; Osborn, T.C. Analysis of gene expression in resynthesized Brassica napus allopolyploids using Arabidopsis 70mer oligo microarrays. PLoS ONE 2009, 4, e4760. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Hovav, R.; Faigenboim-Doron, A.; Kadmon, N.; Hu, G.; Zhang, X.; Gallagher, J.P.; Wendel, J.F. A transcriptome profile for developing seed of polyploid cotton. Plant Genome 2015, 8, 15. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Li, Y.; Zhang, Z.; Li, L.; Liu, B. Transcriptome asymmetry in synthetic and natural allotetraploid wheats, revealed by RNA-sequencing. New Phytol. 2016, 209, 1264–1277. [Google Scholar] [CrossRef] [PubMed]
- Havlickova, L.; He, Z.; Wang, L.; Langer, S.; Harper, A.L.; Kaur, H.; Broadley, M.R.; Gegas, V.; Bancroft, I. Validation of an updated associative transcriptomics platform for the polyploid crop species Brassica napus by dissection of the genetic architecture of erucic acid and tocopherol isoform variation in seeds. Plant J. 2018, 93, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Wang, H.; Song, A.; Jiang, J.; Chen, S.; Chen, F. Genomic and transcriptomic alterations following intergeneric hybridization and polyploidization in the Chrysanthemum nankingense × Tanacetum vulgare hybrid and allopolyploid (Asteraceae). Hortic. Res. 2018, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.Y.; Zhao, Q.; Zou, J.; Wang, W.L.; Gao, Y.; Meng, J.L.; Wang, J.B. Characterization and expression patterns of small RNAs in synthesized Brassica hexaploids. Plant Mol. Biol. 2014, 85, 287–299. [Google Scholar] [CrossRef]
- Tian, E.T.; Jiang, Y.F.; Chen, L.L.; Zou, J.; Liu, F.; Meng, J.L. Synthesis of a Brassica trigenomic allohexaploid (B. carinata × B. rapa) de novo and its stability in subsequent generations. Theor. Appl. Genet. 2010, 121, 1431–1440. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Xiao, G.; Zhang, Z.; Xu, Y.; Guan, C.; Chen, S.; Wu, X. The expression of 9 photosynthesis related genes in different growth stages of high oleic rapeseed near-isogenic lines. Crops 2015, 4, 11–15. [Google Scholar]
- Smolikova, G.; Dolgikh, E.; Vikhnina, M.; Frolov, A.; Medvedev, S. Genetic and hormonal regulation of chlorophyll degradation during maturation of seeds with green embryos. Int. J. Mol. Sci. 2017, 18, 1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holdsworth, M.J.; Bentsink, L.; Soppe, W.J. Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 2008, 179, 33–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstead, I.; Donnison, I.; Aubry, S.; Harper, J.; Hortensteiner, S.; James, C.; Mani, J.; Moffet, M.; Ougham, H.; Roberts, L.; et al. Cross-species identification of Mendel’s I locus. Science 2007, 315, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Li, Z.; Yang, L.; Xie, Z.; Chen, J.; Zhang, W.; Liu, T.; Gao, S.; Gao, J.; Zhu, Y.; et al. Non-yellowing2 (NYE2), a close paralog of NYE1, plays a positive role in chlorophyll degradation in Arabidopsis. Mol. Plant 2016, 9, 624–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leprince, O.; Pellizzaro, A.; Berriri, S.; Buitink, J. Late seed maturation: Drying without dying. J. Exp. Bot. 2017, 68, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, M.J.; Tunnacliffe, A. POPP the question: What do LEA proteins do? Trends Plant Sci. 2004, 9, 13–17. [Google Scholar] [CrossRef]
- Bravo-Garcia, A.; Yasumura, Y.; Langdale, J.A. Specialization of the Golden2-like regulatory pathway during land plant evolution. New Phytol. 2009, 183, 133–141. [Google Scholar] [CrossRef]
- Raffaele, S.; Rivas, S.; Roby, D. An essential role for salicylic acid in AtMYB30-mediated control of the hypersensitive cell death program in Arabidopsis. FEBS Lett. 2006, 580, 3498–3504. [Google Scholar] [CrossRef]
- Lu, N.; Roldan, M.; Dixon, R.A. Characterization of two TT2-type MYB transcription factors regulating proanthocyanidin biosynthesis in tetraploid cotton, Gossypium hirsutum. Planta 2017, 246, 323–335. [Google Scholar] [CrossRef]
- Babiychuk, E.; Vandepoele, K.; Wissing, J.; Garcia-Diaz, M.; De Rycke, R.; Akbari, H.; Joubès, J.; Beeckman, T.; Jänsch, L.; Frentzen, M.; et al. Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc. Natl. Acad. Sci. USA 2011, 108, 6674–6679. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Lee, U.; Small, I.; Des Francs-Small, C.C.; Vierling, E. Mutations in an Arabidopsis mitochondrial transcription termination factor-related protein enhance thermotolerance in the absence of the major molecular chaperone HSP101. Plant Cell 2012, 24, 3349–3365. [Google Scholar] [CrossRef] [Green Version]
- Quesada, V.; Sarmiento-Manus, R.; Gonzalez-Bayon, R.; Hricova, A.; Perez-Marcos, R.; Gracia-Martinez, E.; Medina-Ruiz, L.; Leyva-Diaz, E.; Ponc, M.R.; Micol, J.L. Arabidopsis RUGOSA2 encodes an mTERF family member required for mitochondrion, chloroplast and leaf development. Plant J. 2011, 68, 738–753. [Google Scholar] [CrossRef]
- Xu, W.; Grain, D.; Bobet, S.; Le Gourrierec, J.; Thevenin, J.; Kelemen, Z.; Lepiniec, L.; Dubos, C. Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed. New Phytol. 2014, 202, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, M.Y.; Wang, F.; Tang, J.; Xiong, A.S. Genome-wide analysis of Dof family transcription factors and their responses to abiotic stresses in Chinese cabbage. BMC Genomics 2015, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Miroshnichenko, S.; Tripp, J.; Zur, N.U.; Neumann, D.; Conrad, U.; Manteuffel, R. Immunomodulation of function of small heat shock proteins prevents their assembly into heat stress granules and results in cell death at sublethal temperatures. Plant J. 2005, 41, 269–281. [Google Scholar] [CrossRef]
- Kuki, Y.; Ohno, R.; Yoshida, K.; Takumi, S. Heterologous expression of wheat WRKY transcription factor genes transcriptionally activated in hybrid necrosis strains alters abiotic and biotic stress tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2020, 150, 71–79. [Google Scholar] [CrossRef]
- He, Y.J.; Mao, S.S.; Gao, Y.L.; Zhu, L.Y.; Wu, D.M.; Cui, Y.; Li, J.; Qian, W. Genome-wide identification and expression analysis of WRKY transcription factors under multiple stresses in Brassica napus. PLoS ONE 2016, 11, 18. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.A.; Xie, D.Y.; Sharma, S.B. Proanthocyanidins—A final frontier in flavonoid research? New Phytol. 2005, 165, 9–28. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 2016, 57, 405–430. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.Y.; Liu, L.Z.; Chai, Y.R.; Chen, L.; Yang, T.; Jin, M.Y.; Ma, A.F.; Yan, X.Y.; Zhang, Z.S.; Li, J.N. Localization of QTLs for seed color using recombinant inbred lines of Brassica napus in different environments. Genome 2007, 50, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Y.; Hussain, N.; Li, Z.; Wu, D.; Jiang, L. Allelic Variation of BnaC.TT2.a and Its Association with Seed Coat Color and Fatty Acids in Rapeseed (Brassica napus L.). PLoS ONE 2016, 11, e0146661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chen, L.; Hong, M.; Zhang, Y.; Zu, F.; Wen, J.; Yi, B.; Ma, C.; Shen, J.; Tu, J.; et al. A large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PLoS ONE 2012, 7, e44145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, L.; Guo, S.; An, F.; Du, D. Fine Mapping and Whole-Genome Resequencing Identify the Seed Coat Color Gene in Brassica rapa. PLoS ONE 2016, 11, e0166464. [Google Scholar] [CrossRef] [Green Version]
- Padmaja, L.K.; Agarwal, P.; Gupta, V.; Mukhopadhyay, A.; Sodhi, Y.S.; Pental, D.; Pradhan, A.K. Natural mutations in two homoeologous TT8 genes control yellow seed coat trait in allotetraploid Brassica juncea (AABB). Theor. Appl. Genet. 2014, 127, 339–347. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, L.; Lu, H.; Lang, L.; Zhao, N.; Ding, J.; Xu, A. Development of IP and SCAR markers linked to the yellow seed color gene in Brassica juncea L. Breed. Sci. 2016, 66, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acid. Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acid. Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample | Stage | Clean reads | Clean reads (%) |
|---|---|---|---|---|
| B. rapa | RS1 | Full-size | 44,199,458 | 79.60 |
| RS2 | Full-size | 44,227,522 | 82.06 | |
| RS3 | Full-size | 44,335,290 | 82.26 | |
| RM1 | Mature | 44,399,710 | 82.38 | |
| RM2 | Mature | 45,242,772 | 81.48 | |
| RM3 | Mature | 45,174,524 | 83.82 | |
| Brassica hexaploid | HS1 | Full-size | 45,063,192 | 78.83 |
| HS2 | Full-size | 44,846,972 | 83.21 | |
| HS3 | Full-size | 44,429,210 | 80.01 | |
| HM1 | Mature | 44,641,026 | 88.17 | |
| HM2 | Mature | 45,300,322 | 89.47 | |
| HM3 | Mature | 44,793,850 | 88.47 | |
| B. carinata | CS1 | Full-size | 44,761,646 | 80.61 |
| CS2 | Full-size | 45,066,350 | 83.62 | |
| CS3 | Full-size | 44,490,496 | 80.12 | |
| CM1 | Mature | 44,273,224 | 87.44 | |
| CM2 | Mature | 44,552,900 | 88.00 | |
| CM3 | Mature | 45,318,352 | 86.71 |
| KEGG pathway | RS vs. HS | CS vs. HS | RM vs. HM | CM vs. HM |
|---|---|---|---|---|
| Metabolic pathways | 4 (18,227) | 2 (59,265) | 1 (104,614) | 3 (33,689) |
| RNA transport | 3 (8970) | 2 (46,110) | 1 (132,455) | 4 (2561) |
| Biosynthesis of secondary metabolites | 4 (−14,014) | 3 (37,358) | 2 (86,457) | 1 (195,966) |
| Fatty acid biosynthesis | 4 (−1351) | 1 (8954) | 2 (263) | 3 (−489) |
| Photosynthesis | 1 (7784) | 3 (670) | 2 (873) | 4 (85) |
| Photosynthesis—antenna proteins | 1 (45,663) | 3 (617) | 2 (749) | 4 (−1087) |
| Carbon fixation in photosynthetic organisms | 1 (12,260) | 2 (3551) | 3 (2307) | 4 (102) |
| Ribosome | 3 (6016) | 1 (9419) | 4 (4067) | 2 (8488) |
| Carbon metabolism | 1 (15,852) | 3 (7677) | 2 (15,470) | 4 (1305) |
| Plant hormone signal transduction | 4 (−4483) | 2 (2384) | 3 (−533) | 1 (5448) |
| Biosynthesis of amino acids | 2 (9042) | 3 (9019) | 1 (11,309) | 4 (912) |
| Flavonoid biosynthesis | (1338) | (1264) | (1677) | (1357) |
| Isoflavonoid biosynthesis | (−725) | (−18) | (1617) | (532) |
| Protein processing in endoplasmic reticulum | (−15,263) | (−3309) | (124,955) | (−64,763) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Wang, R.; Wang, J. Comprehensive Transcriptomic Analysis for Developing Seeds of a Synthetic Brassica Hexaploid. Plants 2020, 9, 1141. https://doi.org/10.3390/plants9091141
Liu Z, Wang R, Wang J. Comprehensive Transcriptomic Analysis for Developing Seeds of a Synthetic Brassica Hexaploid. Plants. 2020; 9(9):1141. https://doi.org/10.3390/plants9091141
Chicago/Turabian StyleLiu, Zhengyi, Ruihua Wang, and Jianbo Wang. 2020. "Comprehensive Transcriptomic Analysis for Developing Seeds of a Synthetic Brassica Hexaploid" Plants 9, no. 9: 1141. https://doi.org/10.3390/plants9091141