Comparison of Chloroplast Genomes among Species of Unisexual and Bisexual Clades of the Monocot Family Araceae

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction and Sequencing
2.2. Genome Assembly and Annotation
2.3. Characterization, Comparative Analyses and Phylogenetic Inference
3. Results
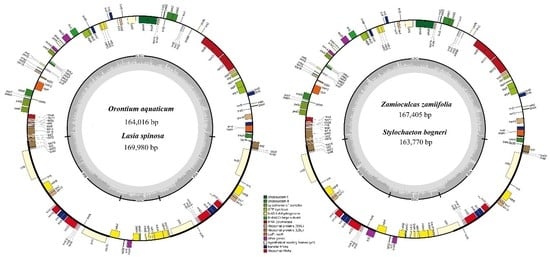
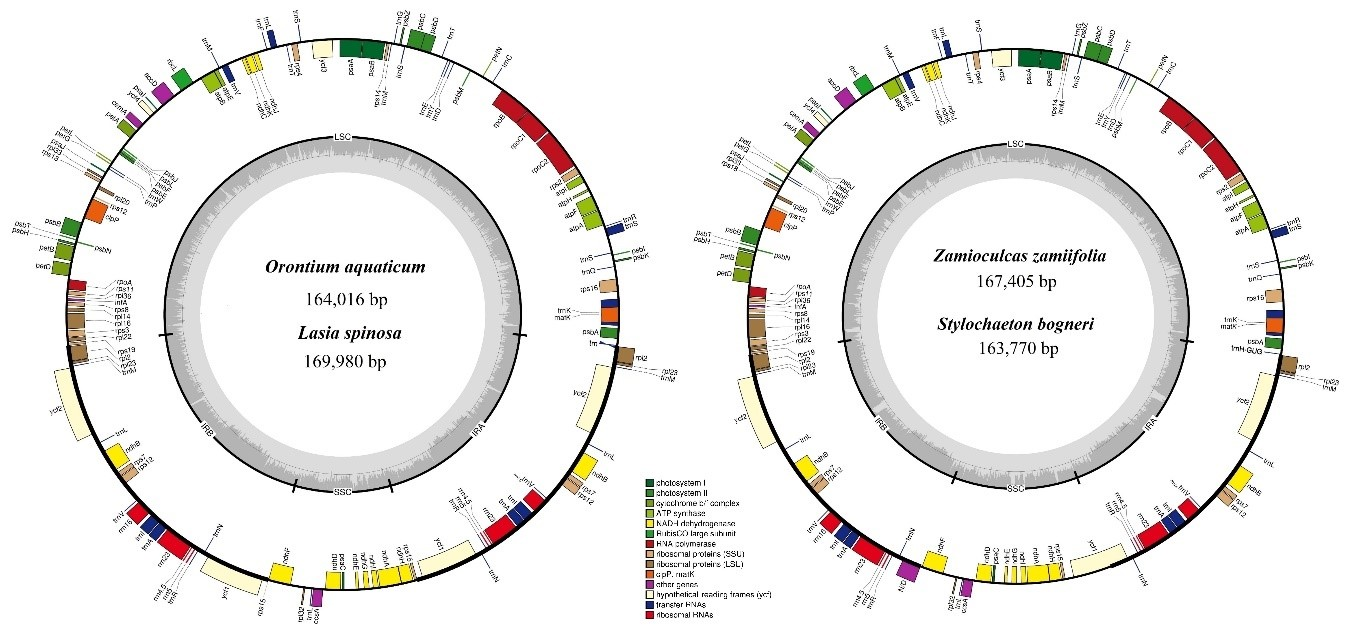
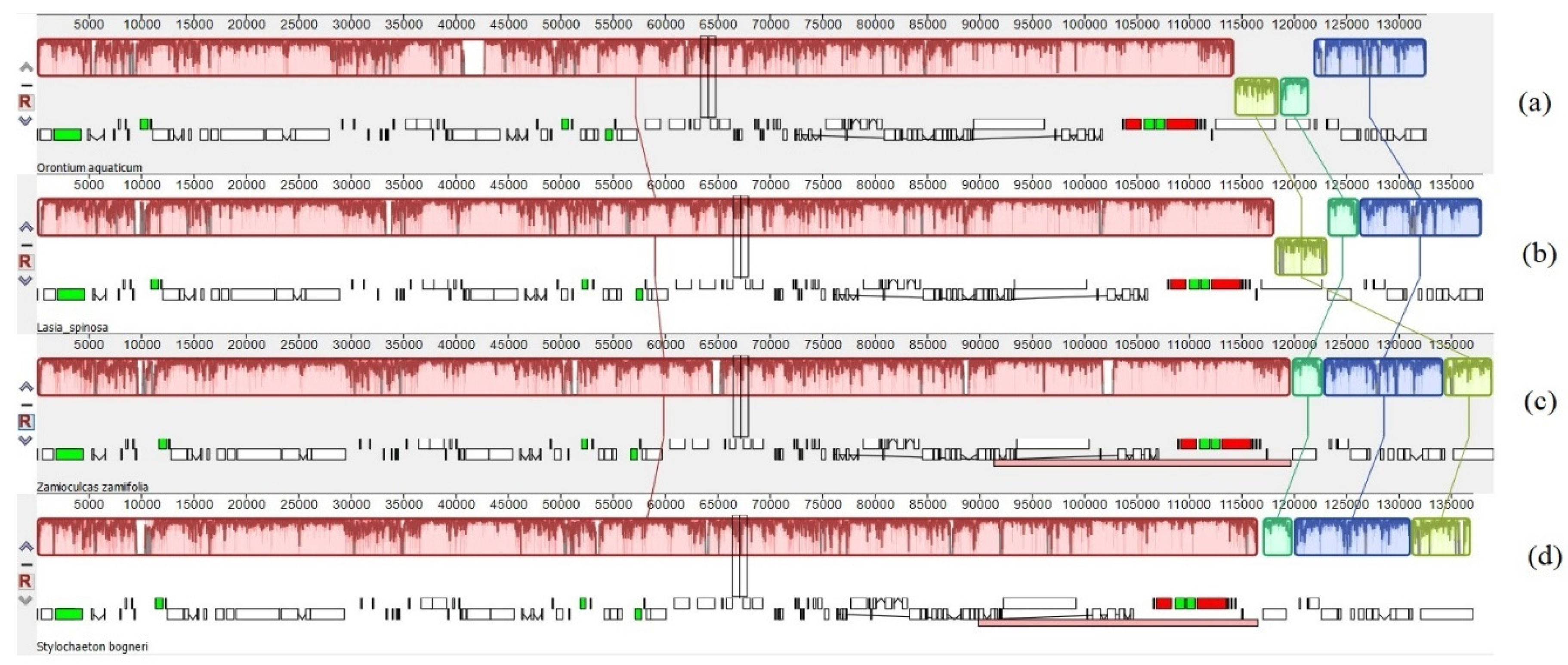
3.1. Comparative Genomics among De Novo Assembled Chloroplast Genomes
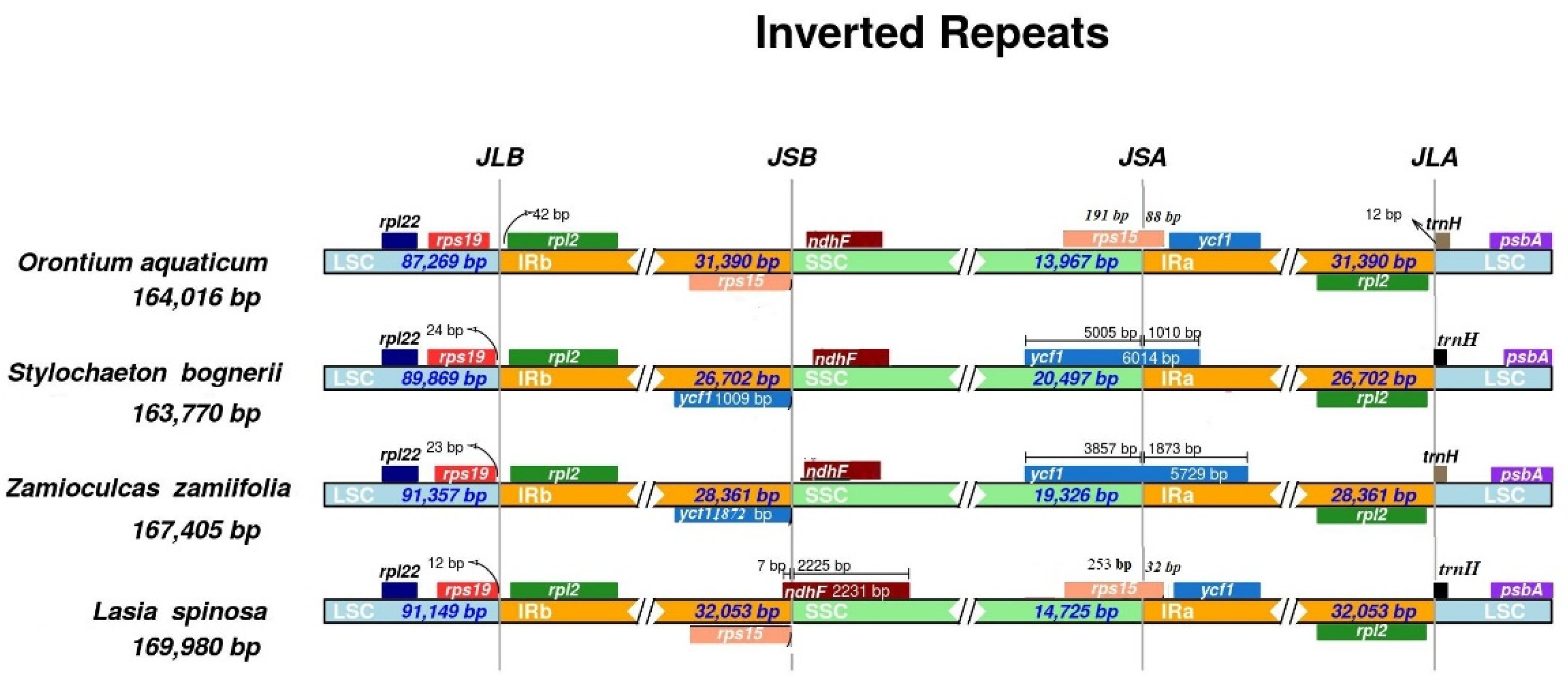
3.2. Contraction and Expansion of Inverted Repeats
3.3. Analyses of Codon Usage, Amino Acid Frequency and RNA Editing
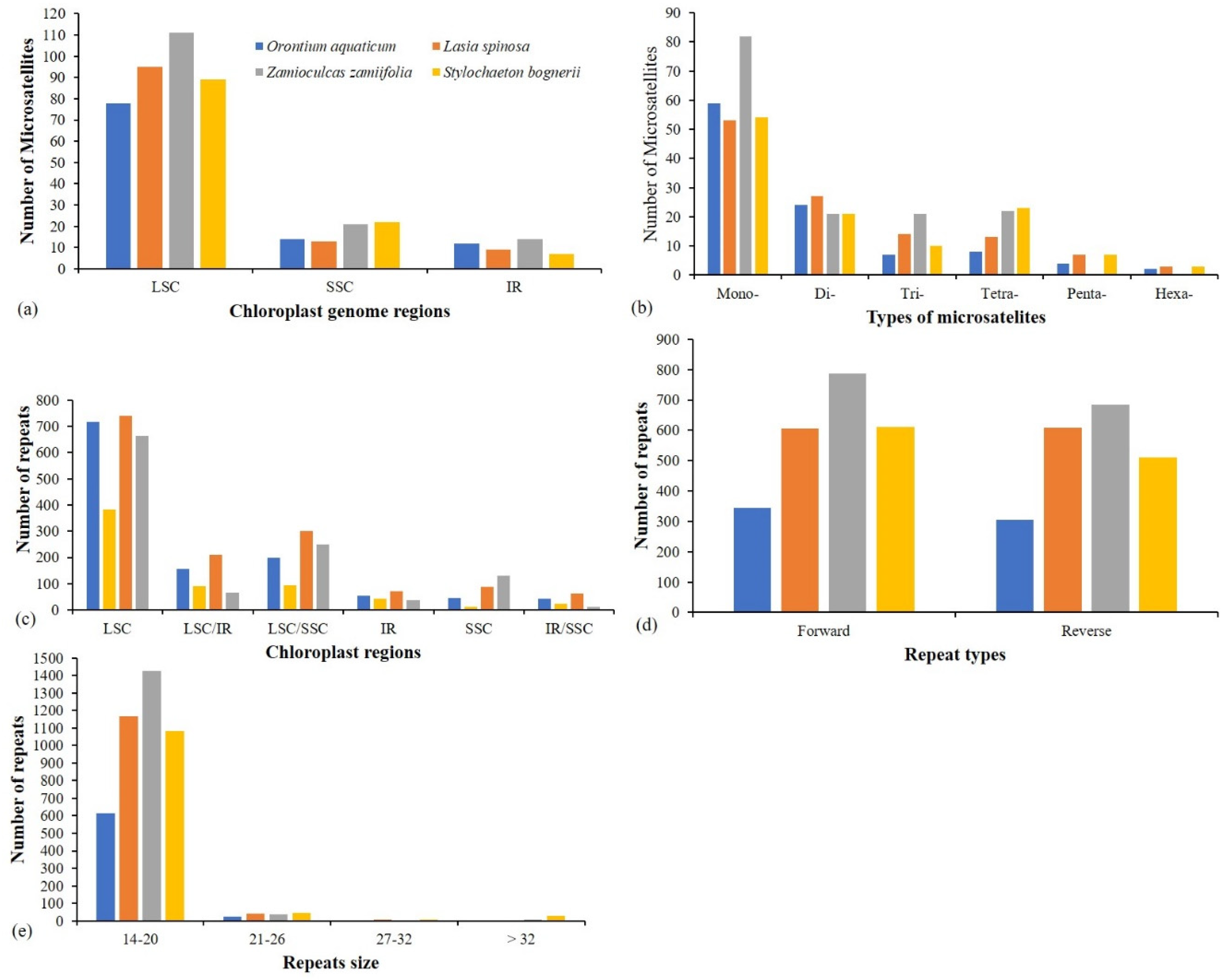
3.4. Repeats Analyses
3.5. Analyses of Substitution Types
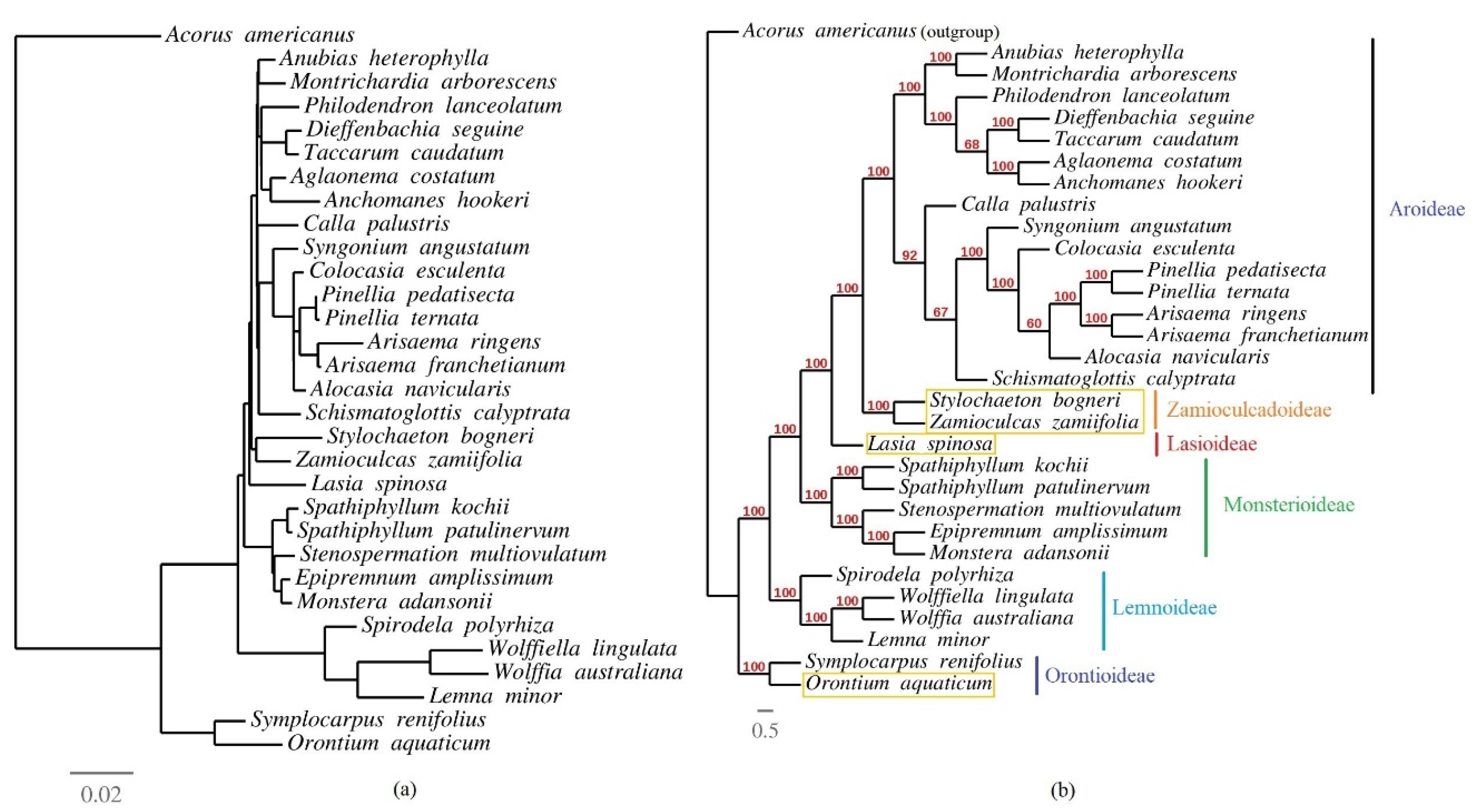
3.6. Phylogenetic Inference of Araceae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, F.; Abdullah; Shahzadi, I.; Ahmed, I.; Waheed, M.T.; Mirza, B. Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 2020, 112, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Abdullah; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genome of Theobroma cacao and Theobroma grandiflorum. Biologia 2020, 75, 761–771. [Google Scholar] [CrossRef]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8, 70. [Google Scholar] [CrossRef]
- Sabir, J.; Schwarz, E.; Ellison, N.; Zhang, J.; Baeshen, N.A.; Mutwakil, M.; Jansen, R.; Ruhlman, T. Evolutionary and biotechnology implications of plastid genome variation in the inverted-repeat-lacking clade of legumes. Plant Biotechnol. J. 2014, 12, 743–754. [Google Scholar] [CrossRef]
- Zeb, U.; Dong, W.; Zhang, T.; Wang, R.; Shahzad, K.; Ma, X.; Li, Z. Comparative plastid genomics of Pinus species: Insights into sequence variations and phylogenetic relationships. J. Syst. Evol. 2019, 12492. [Google Scholar] [CrossRef]
- Abdullah; Henriquez, C.L.; Mehmood, F.; Carlsen, M.M.; Islam, M.; Waheed, M.T.; Poczai, P.; Croat, T.B.; Ahmed, I. Complete chloroplast genomes of Anthurium huixtlense and Pothos scandens (Pothoideae, Araceae): Unique inverted repeat expansion and contraction affect rate of evolution. BioRxiv 2020. [Google Scholar] [CrossRef]
- Oldenburg, D.J.; Bendich, A.J. The linear plastid chromosomes of maize: Terminal sequences, structures, and implications for DNA replication. Curr. Genet. 2016, 62, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Abdullah; Mehmood, F.; Ali, Z.; Ahmed, I.; Mirza, B. Chloroplast genome sequences of Artemisia maritima and Artemisia absinthium: Comparative analyses, mutational hotspots in genus Artemisia and phylogeny in family Asteraceae. Genomics 2020, 112, 1454–1463. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; Mckain, M.R. Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 2020, 112, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Abdullah; Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 2020, 112, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. The chloroplast genome sequence of bittersweet (Solanum dulcamara): Plastid genome structure evolution in Solanaceae. PLoS ONE 2018, 13, 1–23. [Google Scholar] [CrossRef]
- Schwarz, E.N.; Ruhlman, T.A.; Sabir, J.S.M.; Hajrah, N.H.; Alharbi, N.S.; Al-Malki, A.L.; Bailey, C.D.; Jansen, R.K. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in Papilionoids. J. Syst. Evol. 2015, 53, 458–468. [Google Scholar] [CrossRef]
- Rabah, S.O.; Shrestha, B.; Hajrah, N.H.; Sabir, M.J.; Alharby, H.F.; Sabir, M.J.; Alhebshi, A.M.; Sabir, J.S.M.; Gilbert, L.E.; Ruhlman, T.A.; et al. Passiflora plastome sequencing reveals widespread genomic rearrangements. J. Syst. Evol. 2019, 57, 1–14. [Google Scholar] [CrossRef]
- Lopes, A. de S.; Pacheco, T.G.; dos Santos, K.G.; do N. Vieira, L.; Guerra, M.P.; Nodari, R.O.; de Souza, E.M.; de O. Pedrosa, F.; Rogalski, M. The Linum usitatissimum L. plastome reveals atypical structural evolution, new editing sites, and the phylogenetic position of Linaceae within Malpighiales. Plant Cell Rep. 2018, 37, 307–328. [Google Scholar] [CrossRef]
- Abdullah; Shahzadi, I.; Mehmood, F.; Ali, Z.; Malik, M.S.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Comparative analyses of chloroplast genomes among three Firmiana species: Identification of mutational hotspots and phylogenetic relationship with other species of Malvaceae. Plant Gene 2019, 19, 100199. [Google Scholar] [CrossRef]
- Liu, E.; Yang, C.; Liu, J.; Jin, S.; Harijati, N.; Hu, Z.; Diao, Y.; Zhao, L. Comparative analysis of complete chloroplast genome sequences of four major Amorphophallus species. Sci. Rep. 2019, 9, 809. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; Mckain, M.R. Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 2020, 251, 72. [Google Scholar] [CrossRef]
- Iram, S.; Hayat, M.Q.; Tahir, M.; Gul, A.; Abdullah; Ahmed, I. Chloroplast genome sequence of Artemisia scoparia: Comparative analyses and screening of mutational hotspots. Plants 2019, 8, 476. [Google Scholar] [CrossRef]
- Poczai, P.; Hyvönen, J. The complete chloroplast genome sequence of the CAM epiphyte Spanish moss (Tillandsia usneoides, Bromeliaceae) and its comparative analysis. PLoS ONE 2017, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; Sederoff, R.R. Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in Loblolly pine. Theor. Appl. Genet. 1989, 77, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H. Transgene containment by maternal inheritance: Effective or elusive? Proc. Natl. Acad. Sci. USA 2007, 104, 6879–6880. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I. Evolutionary Dynamics in Taro; Massey University: Palmerston North, New Zealand, 2014. [Google Scholar]
- Li, L.-F.; Wang, H.-Y.; Zhang, C.; Wang, X.-F.; Shi, F.-X.; Chen, W.-N.; Ge, X.-J. Origins and Domestication of Cultivated Banana Inferred from Chloroplast and Nuclear Genes. PLoS ONE 2013, 8, e80502. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; Mclenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Feng, L.; Yue, M.; He, Y.L.; Zhao, G.F.; Li, Z.H. Species delimitation and interspecific relationships of the endangered herb genus Notopterygium inferred from multilocus variations. Mol. Phylogenet. Evol. 2019, 133, 142–151. [Google Scholar] [CrossRef]
- Zhai, W.; Duan, X.; Zhang, R.; Guo, C.; Li, L.; Xu, G.; Shan, H.; Kong, H.; Ren, Y. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol. Phylogenet. Evol. 2019, 135, 12–21. [Google Scholar] [CrossRef]
- Mehmood, F.; Abdullah; Ubaid, Z.; Bao, Y.; Poczai, P.; Mirza, B. Comparative plastomics of Ashwagandha (Withania, Solanaceae) and identification of mutational hotspots for barcoding medicinal plants. Preprints 2020. [Google Scholar] [CrossRef]
- Palhares, R.M.; Drummond, M.G.; Dos Santos Alves Figueiredo Brasil, B.; Cosenza, G.P.; Das Graças Lins Brandão, M.; Oliveira, G. Medicinal plants recommended by the world health organization: DNA barcode identification associated with chemical analyses guarantees their quality. PLoS ONE 2015, 10, 1–29. [Google Scholar] [CrossRef]
- Suzuki, J.Y.; Geib, S.M.; Carlsen, M.M.; Henriquez, C.L.; Amore, T.D.; Sim, S.B.; Matsumoto, T.K.; Keith, L.M.; Myers, R.Y. Development of chloroplast single nucleotide polymorphisms (SNPs) as a tool towards interspecies typing of Anthurium germplasm. In Proceedings of the Acta Horticulturae, International Society for Horticultural Science, Krabi, Thailand, 7–9 March 2017; Volume 1167, pp. 257–270. [Google Scholar]
- Boyce, P.C.; Croat, T.B. The Überlist of Araceae, Totals for Published and Estimated Number of Species in Aroid Genera. 2018. Available online: http://www.aroid.org/genera/180211uberlist.pdf (accessed on 9 April 2020).
- Cusimano, N.; Bogner, J.; Mayo, S.J.; Boyce, P.C.; Wong, S.Y.; Hesse, M.; Hetterscheid, W.L.A.; Keating, R.C.; French, J.C. Relationships within the Araceae: Comparison of morphological patterns with molecular phylogenies. Am. J. Bot. 2011, 98, 654–668. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Arias, T.; Pires, J.C.; Croat, T.B.; Schaal, B.A. Phylogenomics of the plant family Araceae. Mol. Phylogenet. Evol. 2014, 75, 91–102. [Google Scholar] [CrossRef]
- Nauheimer, L.; Metzler, D.; Renner, S.S. Global history of the ancient monocot family Araceae inferred with models accounting for past continental positions and previous ranges based on fossils. New Phytol. 2012, 195, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Messing, J. High-Throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Zhu, G.F.; Li, D.M.; Wang, X.J. The complete chloroplast genome sequence of Spathiphyllum cannifolium. Mitochondrial DNA Part B Resour. 2019, 4, 1822–1823. [Google Scholar] [CrossRef]
- Han, L.; Wang, B.; Wang, Z.Z. The complete chloroplast genome sequence of Spathiphyllum kochii. Mitochondrial DNA 2016, 27, 2973–2974. [Google Scholar] [CrossRef]
- Ahmed, I.; Biggs, P.J.; Matthews, P.J.; Collins, L.J.; Hendy, M.D.; Lockhart, P.J. Mutational dynamics of aroid chloroplast genomes. Genome Biol. Evol. 2012, 4, 1316–1323. [Google Scholar] [CrossRef]
- Han, L.; Chen, C.; Wang, B.; Wang, Z.-Z. The complete chloroplast genome sequence of medicinal plant Pinellia ternata. Mitochondrial DNA. Part A, DNA mapping, Seq. Anal. 2016, 27, 2921–2922. [Google Scholar] [CrossRef]
- Choi, K.S.; Park, K.T.; Park, S. The Chloroplast Genome of Symplocarpus renifolius: A comparison of chloroplast genome structure in Araceae. Genes 2017, 8, 324. [Google Scholar] [CrossRef]
- Kim, S.-H.; Yang, J.; Park, J.; Yamada, T.; Maki, M.; Kim, S.-C. Comparison of Whole Plastome Sequences between Thermogenic Skunk Cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 2019, 20, 4678. [Google Scholar] [CrossRef]
- Mayo, S.J.; Bogner, J.; Catherine, E.; Boyce, P.J. The genera of Araceae; Royal Botanic Gardens, Kew: London, UK, 1997; ISBN 9781900347228. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 September 2019).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Abdullah; Mehmood, F.; Shahzadi, I.; Ali, Z.; Islam, M.; Naeem, M.; Mirza, B.; Lockhart, P.; Ahmed, I.; Waheed, M.T. Correlations among oligonucleotide repeats, nucleotide substitutions and insertion-deletion mutations in chloroplast genomes of plant family Malvaceae. J. Syst. Evol. 2020. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet-next generation sequence assembly visualization. Bioinformatics 2009, 26, 401–402. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lehwark, P.; Greiner, S. GB2sequin-A file converter preparing custom GenBank files for database submission. Genomics 2019, 111, 759–761. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Kwak, M.; Lee, B.; Park, S.J. Complete chloroplast genome of Tetragonia tetragonioides: Molecular phylogenetic relationships and evolution in caryophyllales. PLoS ONE 2018, 13, 1–11. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop, GCE 2010, New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: Rearrangements, repeats, and codon usage. Mol. Biol. Evol. 2011, 28, 583–600. [Google Scholar] [CrossRef]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef]
- Mehmood, F.; Abdullah; Ubaid, Z.; Shahzadi, I.; Ahmed, I.; Waheed, M.T.; Poczai, P.; Mirza, B. Plastid genomics of Nicotiana (Solanaceae): Insights into molecular evolution, positive selection and the origin of the maternal genome of Aztec tobacco (Nicotiana rustica). BioRxiv 2020. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; de Pamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef]
- Wikstro, N.; Savolainen, V.; Chase, M.W. Evolution of the angiosperms: Calibrating the family tree. Proc. Biol. Sci. 2001, 268, 2211–2220. [Google Scholar] [CrossRef]
- Gustafsson, A.L.S.; Verola, C.F.; Antonelli, A. Reassessing the temporal evolution of orchids with new fossils and a Bayesian relaxed clock, with implications for the diversification of the rare South American genus Hoffmannseggella (Orchidaceae: Epidendroideae). BMC Evol. Biol. 2010, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Biswal, D.; Konhar, R.; Debnath, M.; Parameswaran, S.; Sundar, D.; Tandon, P. Chloroplast genome sequence annotation of Dendrobium nobile (Asparagales: Orchidaceae), an endangered medicinal Orchid from Northeast India. PLoS Curr. 2017, 9. [Google Scholar] [CrossRef]
- Keller, J.; Rousseau-Gueutin, M.; Martin, G.E.; Morice, J.; Boutte, J.; Coissac, E.; Ourari, M.; Aïnouche, M.; Salmon, A.; Cabello-Hurtado, F.; et al. The evolutionary fate of the chloroplast and nuclear rps16 genes as revealed through the sequencing and comparative analyses of four novel legume chloroplast genomes from Lupinus. DNA Res. 2017, 24, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Han, L.; Chen, C.; Wang, Z. The complete chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PLoS ONE 2018, 13, e0192956. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.J.; Wang, W.C.; Da Huang, H.; Leu, J.Y. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Guisinger, M.; Kim, H.-G.; Ruck, E.; Blazier, J.C.; McMurtry, V.; Kuehl, J.V.; Boore, J.; Jansen, R.K. Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 2008, 67, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef]
- Goulding, S.E.; Olmstead, R.G.; Morden, C.W.; Wolfe, K.H. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Wang, Y.H.; Qu, X.J.; Chen, S.Y.; Li, D.Z.; Yi, T.S. Plastomes of Mimosoideae: Structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet. Genomes 2017, 13, 1–18. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; He, P.; Li, P.; Lee, J.; Soltis, D.E.; Fu, C. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics 2018, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, D.-F.; Jia, X.; Mei, W.-L.; Dai, H.-F.; Chen, X.-T.; Peng, S.-Q. Complete Chloroplast Genome Sequence of Aquilaria sinensis (Lour.) Gilg and Evolution Analysis within the Malvales Order. Front. Plant Sci. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Ng, W.L.; Mohamed, R.; Terhem, R. The complete chloroplast genome of Aquilaria malaccensis Lam. (Thymelaeaceae), an important and threatened agarwood-producing tree species. Mitochondrial DNA Part B 2018, 3, 1120–1121. [Google Scholar] [CrossRef]
- Piot, A.; Hackel, J.; Christin, P.A.; Besnard, G. One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 2018, 247, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zuo, L.; Lu, D.; Lu, B.; Yang, M.; Wang, J. Comparative analysis of chloroplast genomes of five Robinia species: Genome comparative and evolution analysis. Gene 2019, 689, 141–151. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Data in GB | Whole Genome Reads (millions) | Phred Score | Chloroplast Reads (millions) | Average Coverage | Maximum Coverage | NCBI Accession |
|---|---|---|---|---|---|---|---|
| Orontium aquaticum | 5.43 | 20.86 | 37.2 | 0.20 | 124.8 | 1347 | MT226773 |
| Lasia spinosa | 8.35 | 32.00 | 37.6 | 1.74 | 1021 | 1929 | MT226772 |
| Zamioculcas zamiifolia | 11.3 | 43.29 | 35.69 | 1.29 | 774.4 | 1293 | MT226775 |
| Stylochaeton bogneri | 3.31 | 12.71 | 37.39 | 0.15 | 92.7 | 749 | MT226774 |
| Characteristic | O. aquaticum | L. spinosa | Z. zamiifolia | S. bogneri | |
|---|---|---|---|---|---|
| Size (base pair; bp) | 164,016 | 169,980 | 167,405 | 163,770 | |
| LSC length (bp) | 87,269 | 91,150 | 91,357 | 89,869 | |
| SSC length (bp) | 13,967 | 18,551 | 19,326 | 20,497 | |
| IR length (bp) | 31,390 | 32,053 | 28,361 | 26,702 | |
| Number of genes* | 131 (113) | 131 (113) | 130 (113) | 130 (113) | |
| Protein-coding genes* | 85 (79) | 85 (79) | 84 (79) | 84 (79) | |
| tRNA genes* | 37 (30) | 37 (30) | 37 (30) | 37 (30) | |
| rRNA genes* | 8 (4) | 8 (4) | 8 (4) | 8 (4) | |
| Duplicate genes | 18 | 18 | 17 | 17 | |
| GC content | Total (%) | 37.3 | 36.1 | 35.9 | 35.7 |
| LSC (%) | 35.7 | 33.9 | 34.2 | 34.0 | |
| SSC (%) | 31.9 | 31.0 | 30.4 | 29.5 | |
| IR (%) | 40.6 | 41.8 | 39.7 | 40.5 | |
| CDS (%) | 37.7 | 37.8 | 37.5 | 37.9 | |
| rRNA (%) | 55.2 | 54.6 | 55.0 | 55.0 | |
| tRNA (%) | 53.1 | 52.9 | 53.2 | 53.1 | |
| All gene % | 39.2 | 39.3 | 39.0 | 39.2 | |
| Substitution Type | Lasia spinosa | Zamioculcas zamiifolia | Stylochaeton bogneri |
|---|---|---|---|
| A/C | 479 | 467 | 562 |
| C/T | 1376 | 1269 | 1459 |
| A/G | 1432 | 1316 | 1543 |
| A/T | 275 | 254 | 316 |
| C/G | 160 | 156 | 195 |
| G/T | 305 | 322 | 407 |
| Ts/Tv | 2.3 | 2.15 | 2.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdullah; Henriquez, C.L.; Mehmood, F.; Shahzadi, I.; Ali, Z.; Waheed, M.T.; Croat, T.B.; Poczai, P.; Ahmed, I. Comparison of Chloroplast Genomes among Species of Unisexual and Bisexual Clades of the Monocot Family Araceae. Plants 2020, 9, 737. https://doi.org/10.3390/plants9060737
Abdullah, Henriquez CL, Mehmood F, Shahzadi I, Ali Z, Waheed MT, Croat TB, Poczai P, Ahmed I. Comparison of Chloroplast Genomes among Species of Unisexual and Bisexual Clades of the Monocot Family Araceae. Plants. 2020; 9(6):737. https://doi.org/10.3390/plants9060737
Chicago/Turabian StyleAbdullah, Claudia L. Henriquez, Furrukh Mehmood, Iram Shahzadi, Zain Ali, Mohammad Tahir Waheed, Thomas B. Croat, Peter Poczai, and Ibrar Ahmed. 2020. "Comparison of Chloroplast Genomes among Species of Unisexual and Bisexual Clades of the Monocot Family Araceae" Plants 9, no. 6: 737. https://doi.org/10.3390/plants9060737
APA StyleAbdullah, Henriquez, C. L., Mehmood, F., Shahzadi, I., Ali, Z., Waheed, M. T., Croat, T. B., Poczai, P., & Ahmed, I. (2020). Comparison of Chloroplast Genomes among Species of Unisexual and Bisexual Clades of the Monocot Family Araceae. Plants, 9(6), 737. https://doi.org/10.3390/plants9060737