Cytoplasm Types Affect DNA Methylation among Different Cytoplasmic Male Sterility Lines and Their Maintainer Line in Soybean (Glycine max L.)



Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. WGBS Library Construction
2.3. WGBS Data Analysis
2.4. Re-Sequencing of the A Lines and the B Line
3. Results
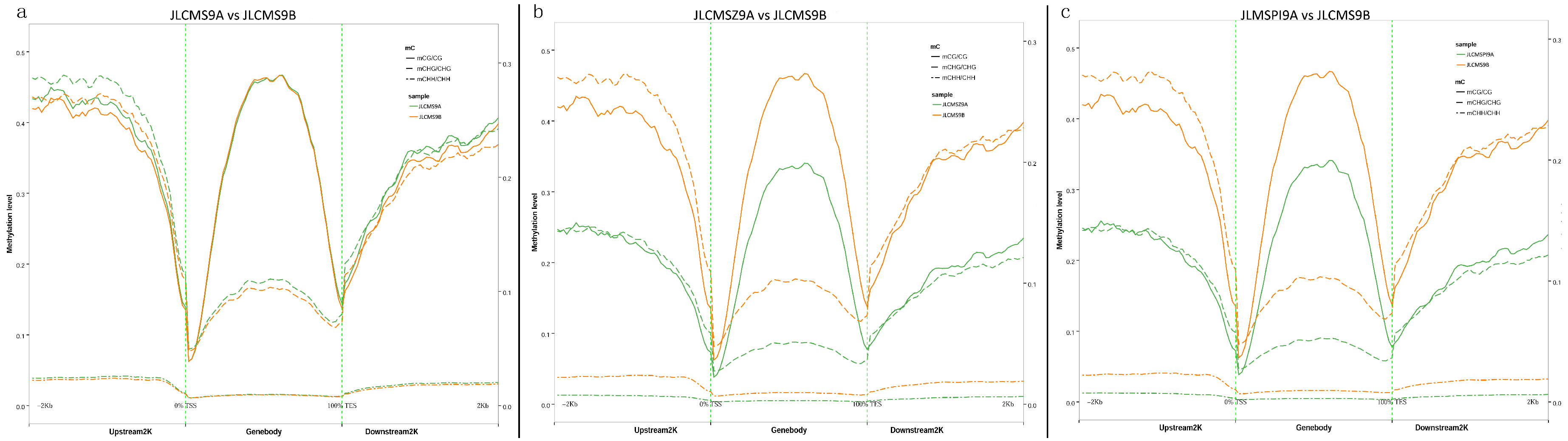
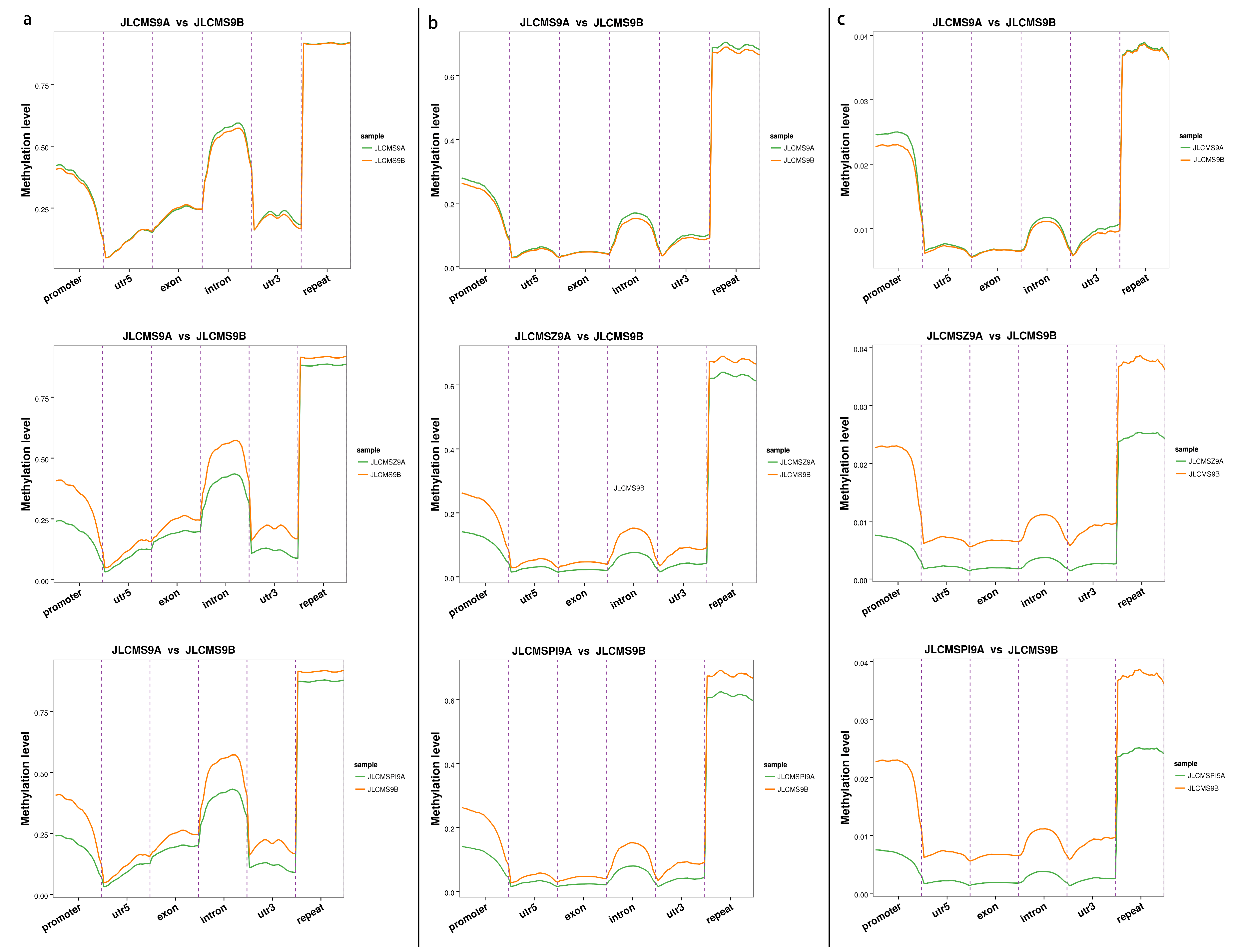
3.1. Whole DNA Methylation Levels of Soybean Lines with Different Cytoplasm types
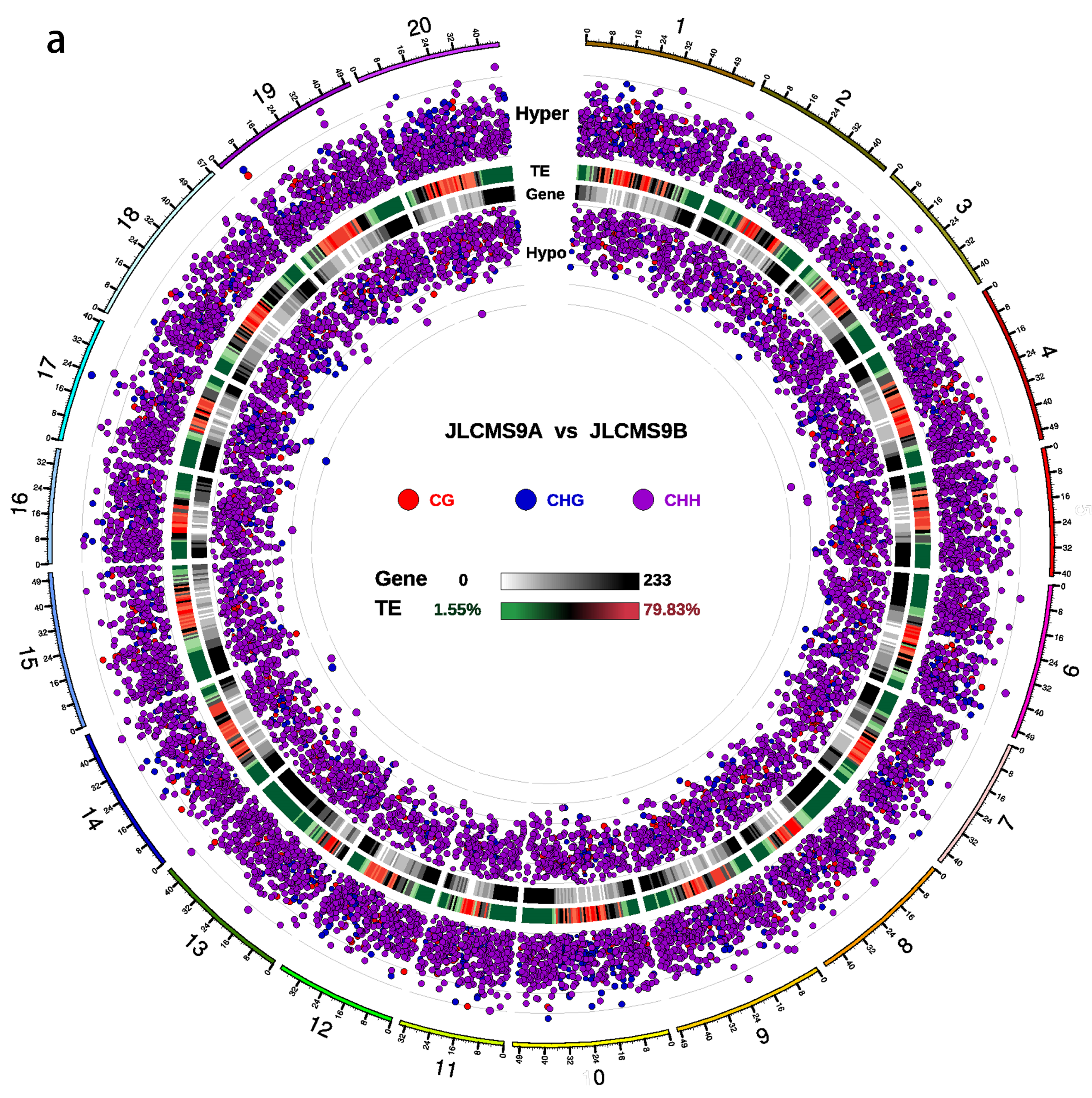
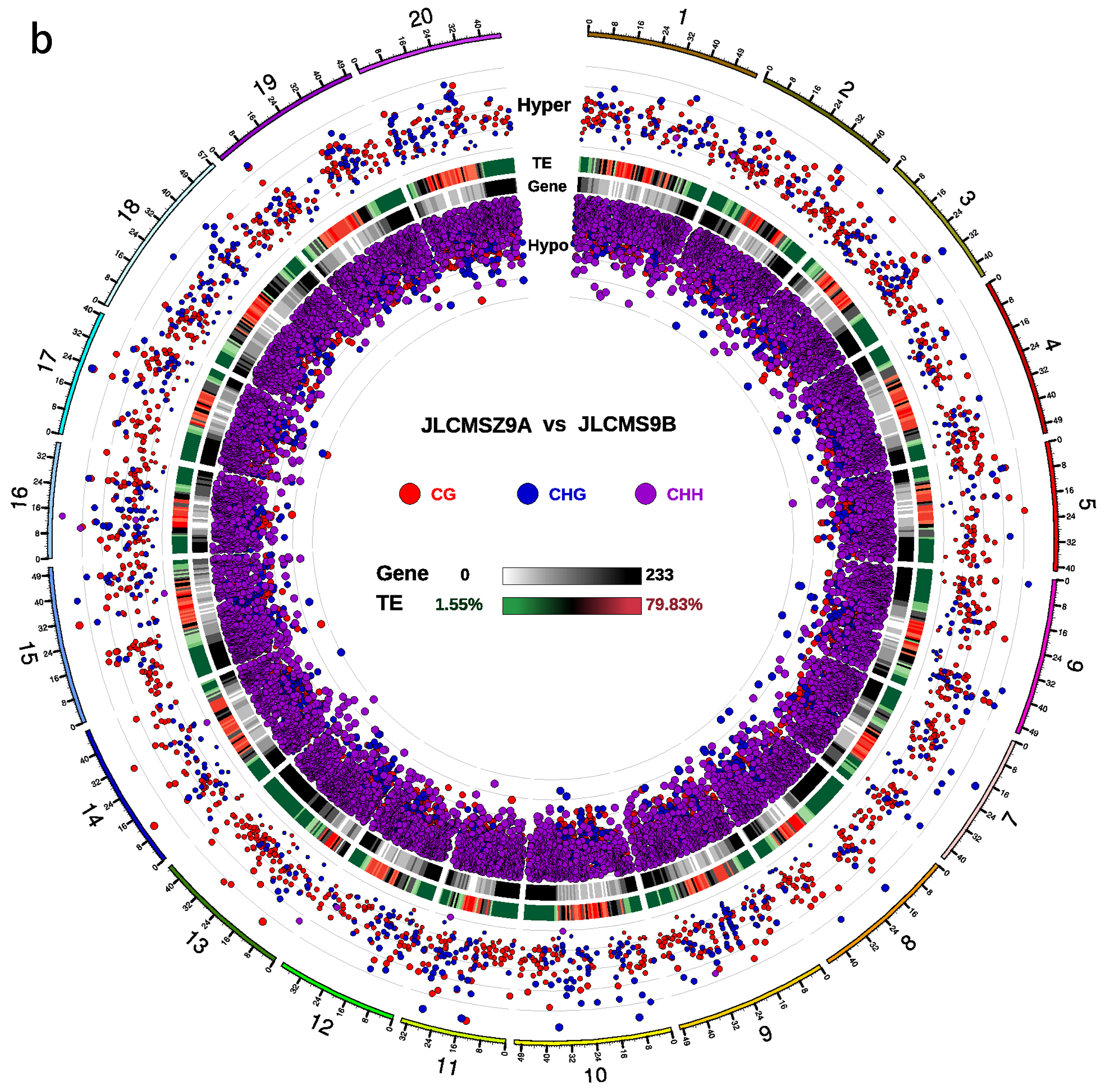
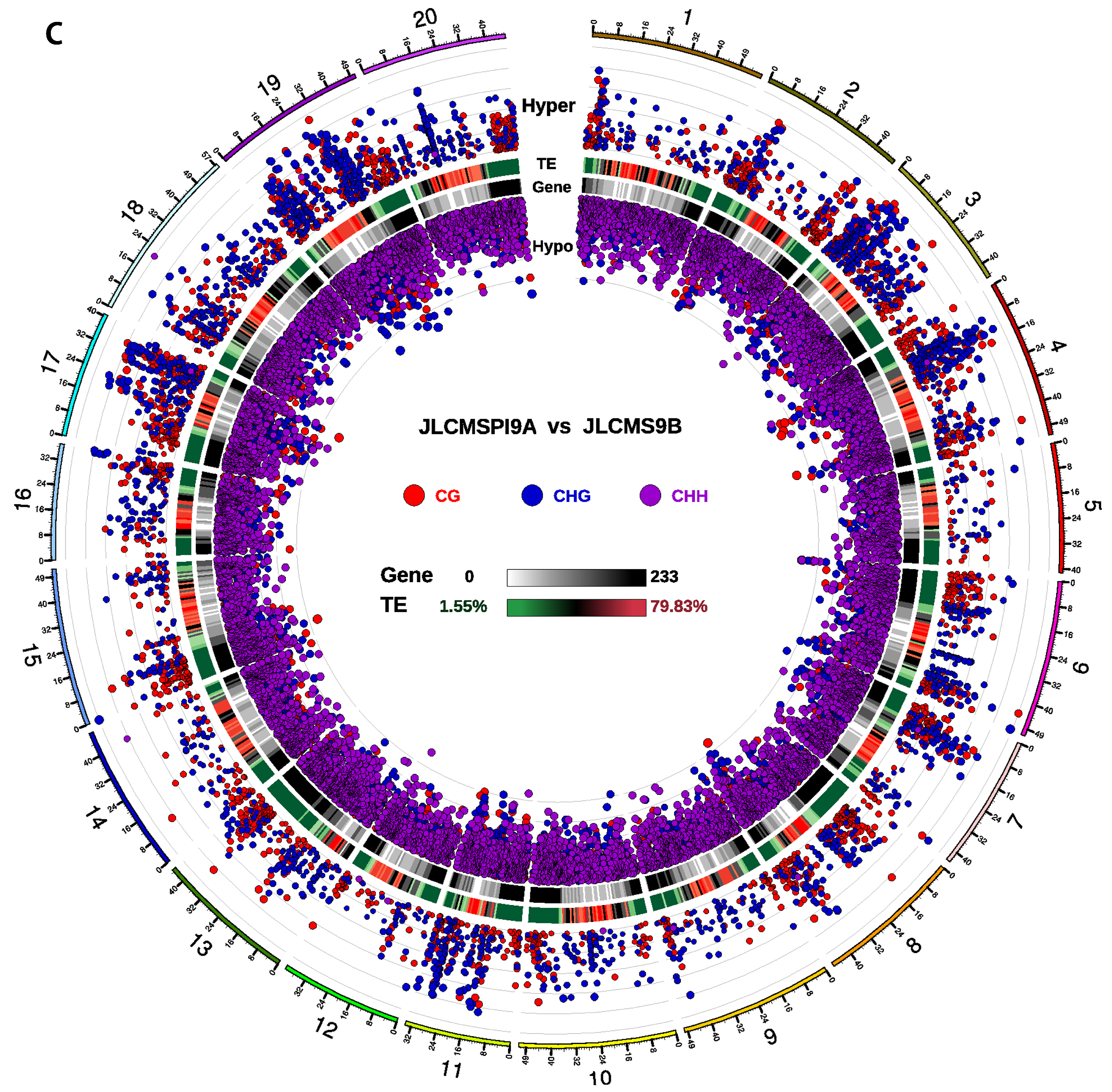
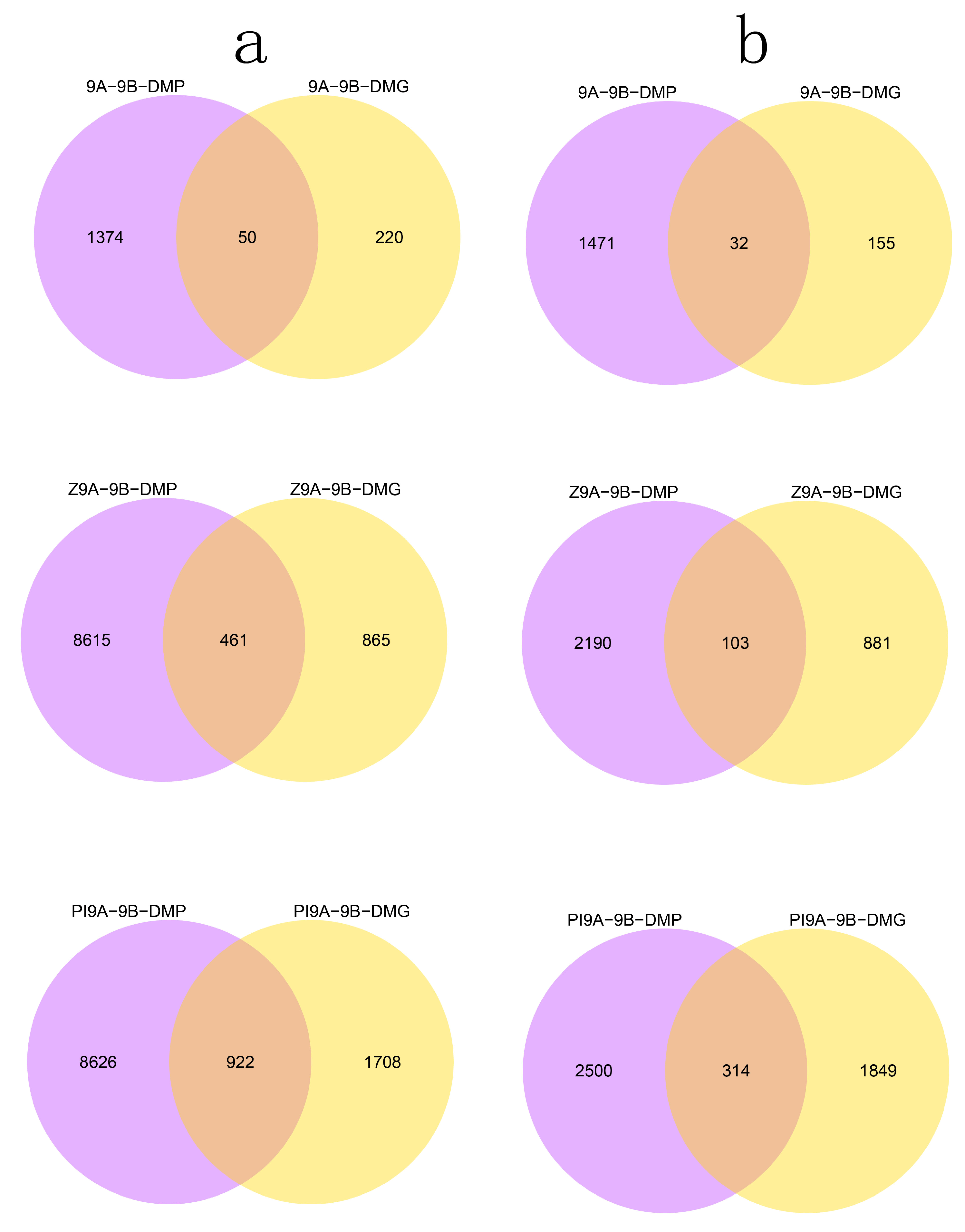
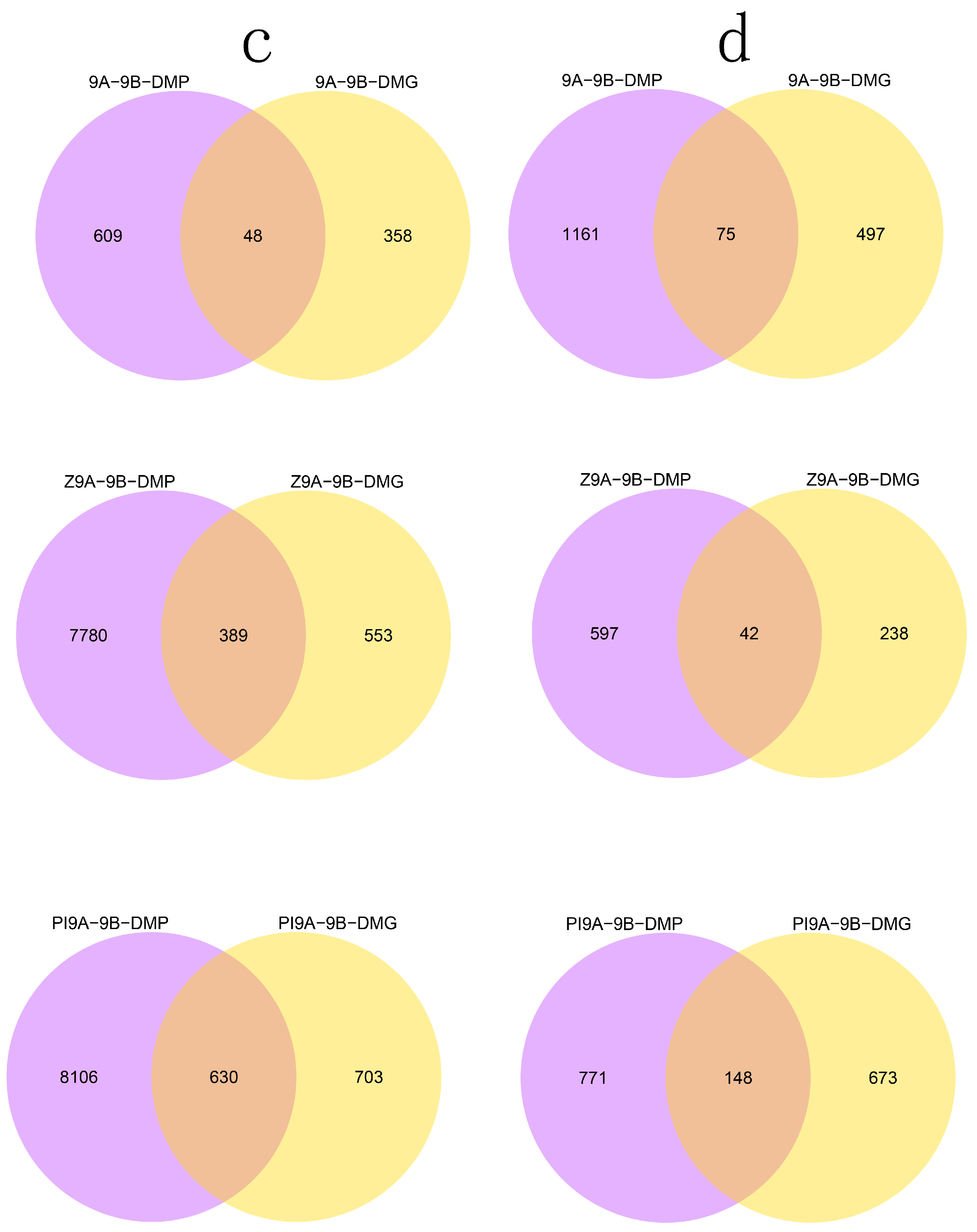
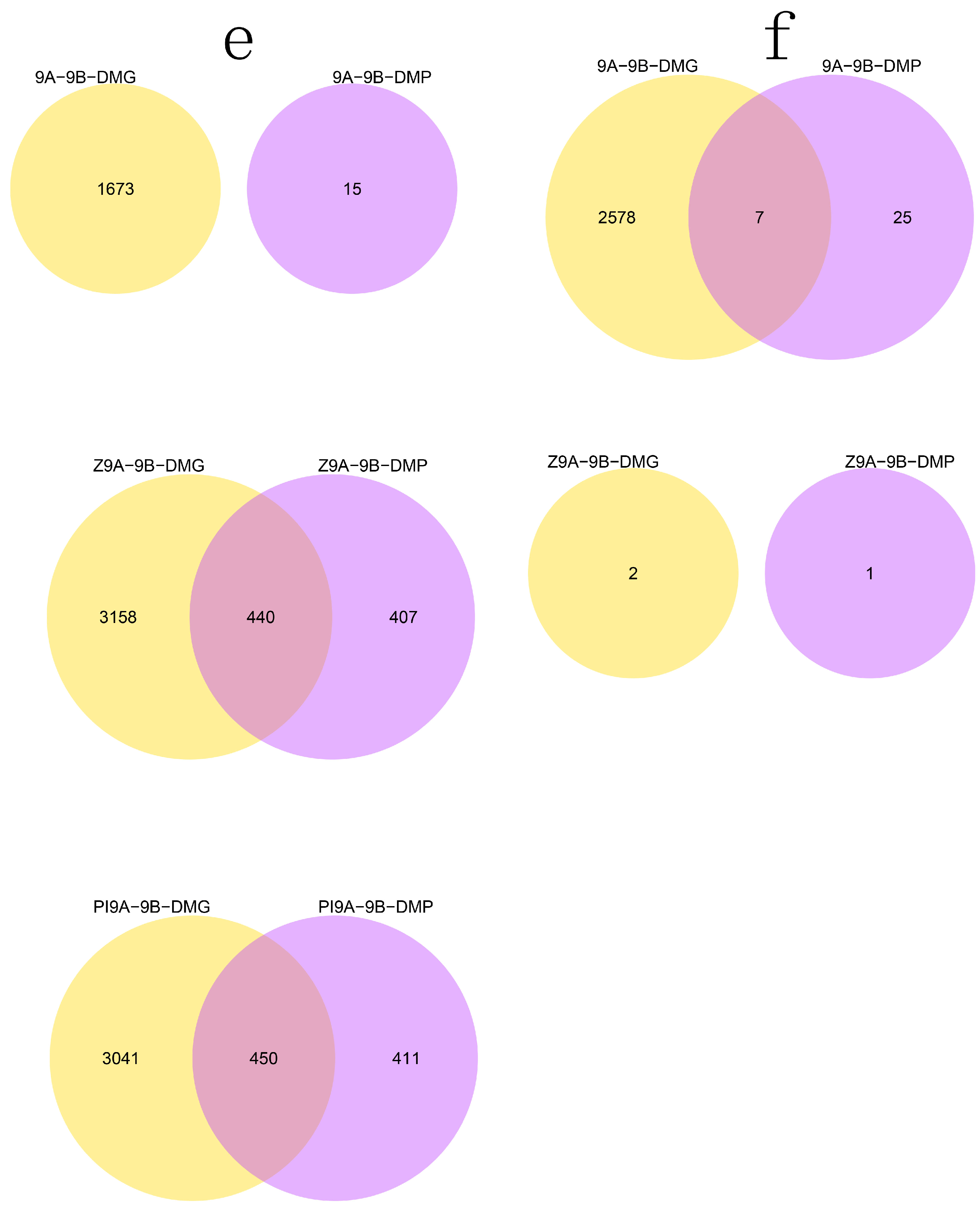
3.2. DMR Analyses in the Three CMS Types
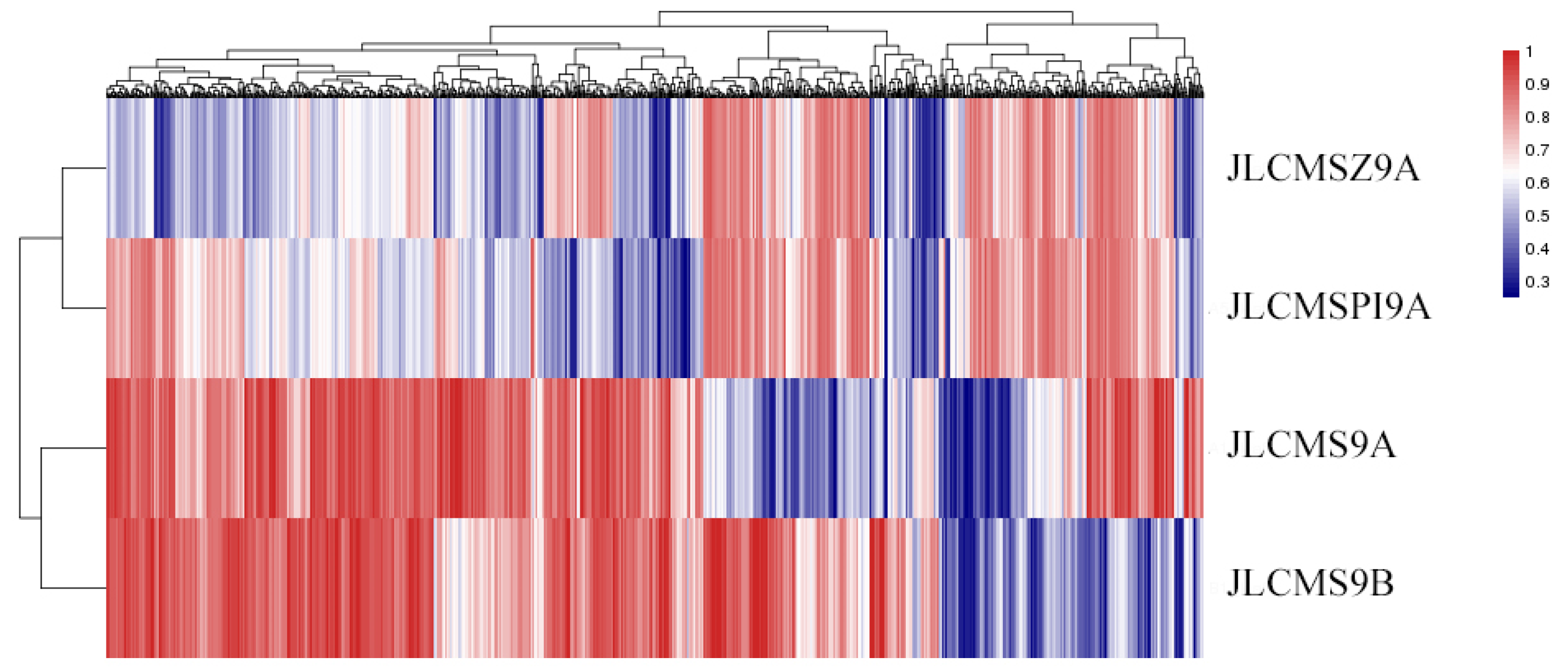
3.3. DNA Methylation Levels and Genetic Distances between Three Soybean CMS Lines and the Maintainer Line
3.4. DMRs Potentially Involved in CMS among the A Lines and B Line in Soybean
4. Discussion
4.1. Cytoplasmic Effects of DNA Methylation among CMS Lines (A line) and Their Maintainer Line (B line)
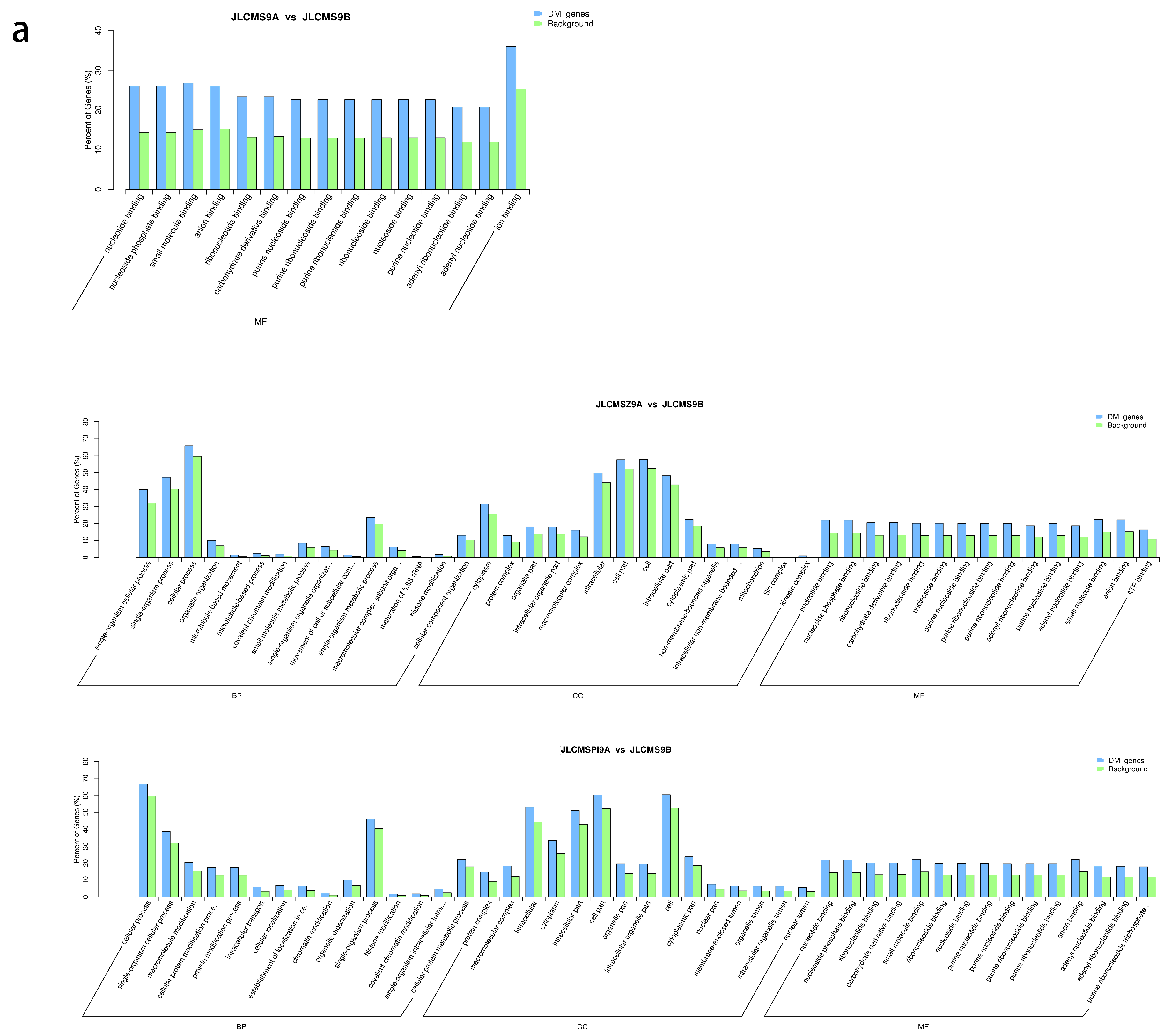
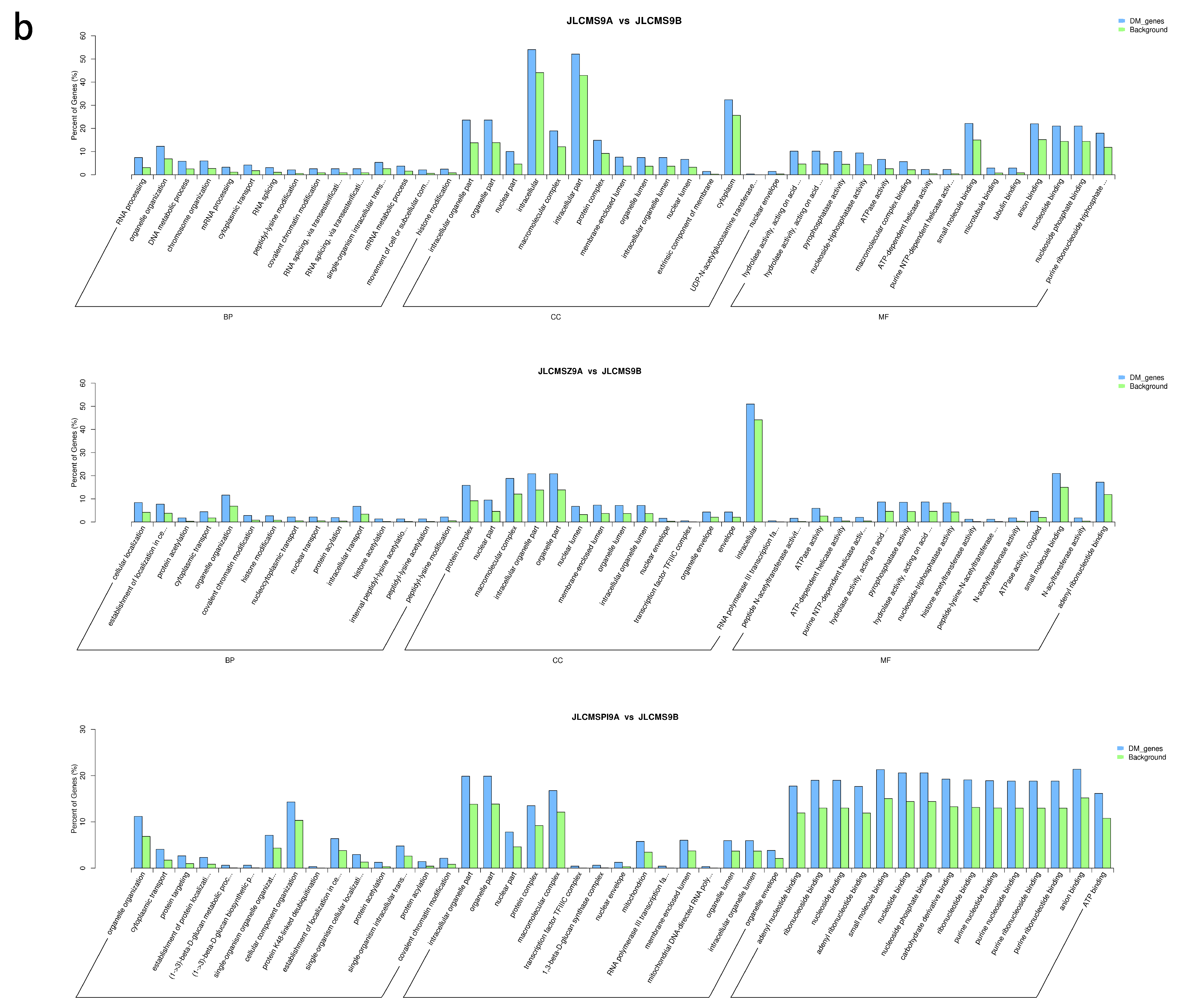
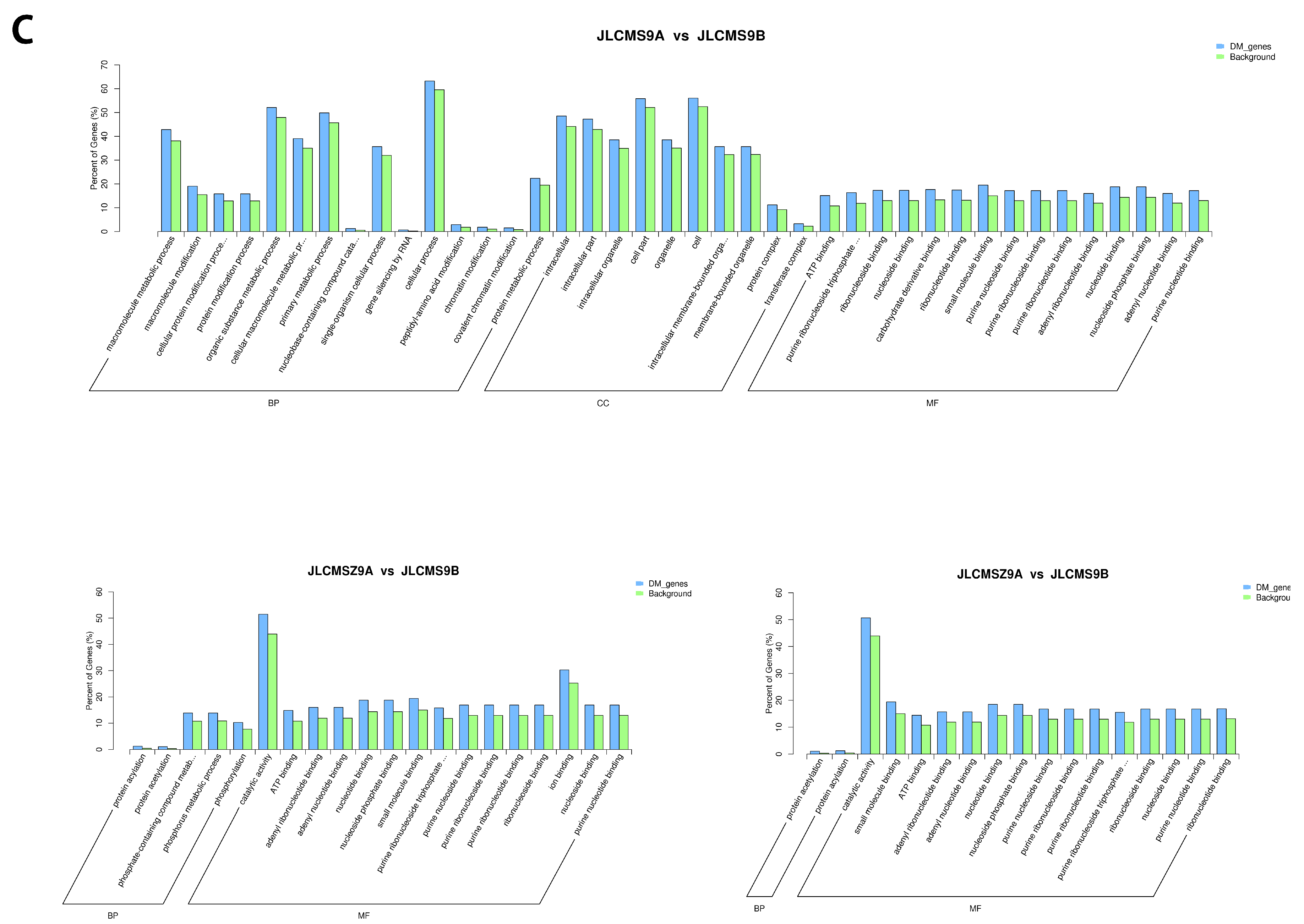
4.2. Enrichment Analysis of DMR-Related Genes (DMGs) Potentially Related to CMS
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CMS | Cytoplasmic male sterility |
| A line | Male sterile lines |
| B line | Maintainer line |
| WGBS | Whole-genome bisulfite sequencing |
| DMR | differentially methylated region |
| MSAP | methylation-sensitive amplified polymorphism |
| DMG | DMR-related gene |
| DMPs | differentially methylated promoter regions |
| mC | Methylated cytosine |
| umC | Unmethylated cytosine |
| Q-PCR | Real-time quantitative PCR |
| ML | methylation level |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes. |
References
- Havey, M. The use of Cytoplasmic Male Sterility for Hybrid-Seed Production. In Molecular Biology and Biotechnology of Plant Organelles: Chloroplasts and Mitochondria; Springer: Dordrecht, The Netherlands, 2004; pp. 623–634. [Google Scholar]
- Zhang, J.; Sun, H.; Zhao, L.; Zhang, C.; Yan, H.; Peng, B.; Li, W. Nectar secretion of RN-type cytoplasmic male sterility three lines in soybean [Glycine max (L.) Merr.]. J. Integr. Agric. 2018, 17, 125–132. [Google Scholar] [CrossRef]
- Davis, W.H. Route to hybrid soybean production. U.S. Patent (Patent No. US4545146), 8 October 1985. [Google Scholar]
- Zhao, L.; Sun, H.; Wang, S.; Wang, Y.; Hang, M.; Li, J. Breeding of hybrid soybean HybSoy. Chin. J. Oil Crop Sci. 2004, 3, 4. [Google Scholar]
- Zhang, L.; Dai, O.; Huang, Z. Selection of soybean male sterile line of nucleo-cytoplasmic interaction and its fertility. Sci. Agric. Sin. 1999, 32, 34–38. [Google Scholar]
- Gai, J.; Cui, Z.; Ji, D.; Ren, Z.; Ding, D. A report on the nuclear cytoplasmic male sterility from a cross between two soybean cultivars. Soybean Genet. Newslett. 1995, 22, 55–58. [Google Scholar]
- Bai, Y.; Gai, J. Development of a new cytoplasmic-nuclear male-sterile line of soybean and inheritance of its male-sterile restoration. Plant Breed 2006, 125, 85–88. [Google Scholar] [CrossRef]
- Zhao, T.; Gai, J. Discovery of new male-sterile cytoplasm sources and development of a new cytoplasmic-nuclear male-sterile line NJCMS3A in soybean. Eupytica 2006, 152, 387–396. [Google Scholar] [CrossRef]
- Li, J.; Nadeem, M.; Sun, G.; Wang, X.; Qiu, L. Male sterility in soybean: Occurrence, molecular basis and utilization. Plant Breeding 2019, 138, 659–676. [Google Scholar] [CrossRef]
- Vertino, P.M.; Yen, R.W.; Gao, J.; Baylin, S.B. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol. Cell Biol. 1996, 16, 4555–4565. [Google Scholar] [CrossRef]
- Deniz, Ö.; Frost, J.; Branco, M.F.; Migurl, R.B. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Jackson, J.P.; Lindroth, A.M.; Cao, X.; Jacobsen, S.E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 2002, 416, 556–560. [Google Scholar] [CrossRef]
- Ba, Q.; Zhang, S.; Niu, N.; Ma, S.; Wang, J. Cytoplasmic effects on DNA methylation between male sterile lines and the maintainer in wheat (Triticum aestivum L.). Gene 2014, 549, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Yan, W.; He, J.; Li, Y.; Zhang, H.; Peng, H.; Wu, X. DNA methylation affected by male sterile cytoplasm in rice (Oryza sativa L.). Mol. Breed. 2013, 31, 719–727. [Google Scholar] [CrossRef]
- Fulneček, J.; Kovařík, A. How to interpret methylation sensitive amplified polymorphism (MSAP) profiles. BMC Genet. 2014, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.B.; Siejka-Zielińska, P.; Velikova, G.; Ying, B.; Fang, Y.; Marketa, T.; Bai, C.; Chen, L.; Schuster-Böckler, B.; Song, C. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Bai, X.; Yang, W.; Wang, W.; Chen, Z.; Ma, J.; Ma, T. Single-base-resolution methylomes of Populus euphratica reveal the association between DNA methylation and salt stress. Tree Genet. Genomes 2018, 14, 86. [Google Scholar] [CrossRef]
- Li, Y.; Ding, X.; Wang, X.; He, T.; Zhang, H.; Yang, L.; Wang, T.; Chen, L.; Gai, J.; Yang, S. Genome-wide comparative analysis of DNA methylation between soybean cytoplasmic male-sterile line NJCMS5A and its maintainer NJCMS5B. BMC Genomics 2017, 18, 596. [Google Scholar] [CrossRef]
- Han, S.; Li, Y.; Li, J.; Zhang, H.; Ding, X.; He, T.; Ga, J.; Yang, S. Genome-wide analysis of DNA methylation to identify genes and pathways associated with male sterility in soybean. Mol. Breed. 2018, 38, 118. [Google Scholar] [CrossRef]
- Sun, H.; Zhao, L.; Huang, M. Studies on cytoplasmic-nuclear male sterile soybean. Chin. Sci. Bull. 1994, 39, 175–176. [Google Scholar]
- Sun, H.; Zhao, L.; Huang, M. ZA Type of cytoplasmic male-sterile breeding and primary research in soybean. Soybean Sci. 1998, 17, 268–270. [Google Scholar]
- Sun, H.; Zhao, L.; Huang, M.; Wang, Y.; Li, J. Advances in utilization of heterosis in soybean. Chin. J. Oil Crop Sci. 2003, 25, 92–96. [Google Scholar]
- Beckmann, J.S.; Osborn, T.C. Plant genomes: Methods for genetic and physical mapping. In Plant Genomes: Methods for Genetic and Physical Mapping; Springer: Dordrecht, The Netherlands, 1992; Volume 85, pp. 331–340. [Google Scholar]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 7271, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.I.; Schein, J.E.; Biro, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, T.; Feng, H.; Chen, L.; Li, B.; Yao, B.; Qin, Z.; Jin, P.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar] [CrossRef]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef]
- Ujjwal, M.; Saurav, M.; Anirban, M.; Sanghamitra, B. Analyzing large gene expression and methylation data profiles using StatBicRM: Statistical Biclustering-Based Rule Mining. PLoS ONE 2015, 10, e0119448. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Cozzetto, D.; Jones, D.T. Computational Methods for Annotation Transfers from Sequence. Methods Mol. Biol. 2016, 1446, 55–67. [Google Scholar]
- Minoru, K.; Susumu, G. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nuleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoke, R.A.; Fennel, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Quantitative analysis of the relationship of biology species using Pearson correlation coefficient. Comput. Eng. Appl. 2005, 41, 79–81. [Google Scholar]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsenet, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Zhao, X.; Chai, Y.; Liu, B. Epigenetic inheritance and variation of DNA methylation level and pattern in maize intra-specific hybrids. Plant Sci. 2007, 172, 930–938. [Google Scholar] [CrossRef]
- Sun, H.; Zhao, L.; Huang, M. Cytoplasmic-genetic male sterile soybean and method for producing hybrid soybean. U.S. Patent (Patent No. US6320098), 20 November 2001. [Google Scholar]
- Chen, X.; Hu, J.; Zhang, H.; Ding, Y. DNA methylation changes in photoperiod-thermo-sensitive male sterile rice PA64S under two different conditions. Gene 2014, 537, 143–148. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.; Li, Q.; Yu, L.; Song, Q.; Gai, J.; Yang, S. Metabolomics studies on cytoplasmic male sterility during flower bud development in soybean. Int. J. Mol. Sci. 2019, 20, 2869. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Clean_Reads | Clean_Bases (Gb) | Q30 * (%) | Unique Mapping Rate (%) | GC Content ** (%) | BS *** Conversion Rate (%) |
|---|---|---|---|---|---|---|
| JLCMS9A | 162450767 | 38.95 | 94.89 | 66.39 | 24.14 | 99.59 |
| JLCMSZ9A | 144302087 | 35.64 | 96.73 | 74.53 | 20.74 | 99.90 |
| JLCMSPI9A | 143848786 | 35.51 | 97.63 | 74.06 | 20.35 | 99.92 |
| JLCM9B | 162499642 | 39.87 | 96.97 | 65.27 | 23.23 | 99.54 |
| Samples | mC * Percent (%) | mCpG ** Percent (%) | mCHG *** Percent (%) | mCHH **** Percent (%) |
|---|---|---|---|---|
| JLCMS9A | 11.27 | 46.36 | 35.66 | 3.29 |
| JLCMSZ9A | 10.16 | 45.77 | 35.88 | 1.91 |
| JLCMSPI9A | 10.48 | 46.65 | 36.75 | 2.08 |
| JLCM9B | 9.76 | 42.14 | 31.8 | 2.47 |
| Samples (A-Line vs B-line) | Gene ID | DMR Region | Type (Site/Level) * | Description |
|---|---|---|---|---|
| JLCMS9A/JLCM9B | GLYMA_09G056300 | Exon | CHH/Hyper | Plasma membrane ATPase 4 |
| JLCMS9A/JLCM9B | GLYMA_13G210600 | Exon | CHH/Hypo | EFTM_ARATH Elongation factor Tu, mitochondrial |
| JLCMS9A/JLCM9B | GLYMA_20G050400 | Exon | CHH/Hypo | MADS-box protein AGL80 |
| JLCMSZ9A/JLCM9B | GLYMA_01G006400 | Exon | CG/Hyper | Arginine--tRNA ligase, chloroplastic/mitochondrial |
| JLCMSZ9A/ JLCM9B | GLYMA_10G132200 | Exon | CHH/Hypo | MYB2_ORYSJ Transcription factor MYB2 |
| JLCMSZ9A/ JLCM9B | GLYMA_10G098600 | Promoter | CG/Hypo | Uncharacterized mitochondrial protein |
| JLCMSZ9A/ JLCM9B | GLYMA_05G068000 | Promoter | CHH/Hypo | Beta-amylase 3 |
| JLCMSZ9A/ JLCM9B | GLYMA_10G111400 | Promoter, exon | CG/Hyper | WRKY transcription factor 42 |
| JLCMSZ9A/ JLCM9B | GLYMA_12G154400 | Exon | CHG/Hyper | Cytochrome c oxidase |
| JLCMSPI9A/ JLCM9B | GLYMA_03G039900 | Promoter, exon | CG/Hyper | Clustered mitochondria protein |
| JLCMSPI9A/ JLCM9B | GLYMA_03G087800 | Promoter | CG/Hyper | WRKY transcription factor |
| JLCMSPI9A/ JLCM9B | GLYMA_07G105800 | Exon | CG/Hyper | Pentatricopeptide repeat-containing protein |
| JLCMSPI9A/ JLCM9B | GLYMA_03G039900 | Promoter, exon | CG/Hyper | Clustered mitochondria protein |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.; Peng, B.; Li, Y.; Wang, P.; Zhao, G.; Ding, X.; Li, R.; Zhao, L.; Zhang, C. Cytoplasm Types Affect DNA Methylation among Different Cytoplasmic Male Sterility Lines and Their Maintainer Line in Soybean (Glycine max L.). Plants 2020, 9, 385. https://doi.org/10.3390/plants9030385
Lin C, Peng B, Li Y, Wang P, Zhao G, Ding X, Li R, Zhao L, Zhang C. Cytoplasm Types Affect DNA Methylation among Different Cytoplasmic Male Sterility Lines and Their Maintainer Line in Soybean (Glycine max L.). Plants. 2020; 9(3):385. https://doi.org/10.3390/plants9030385
Chicago/Turabian StyleLin, Chunjing, Bao Peng, Yongkuan Li, Pengnian Wang, Guolong Zhao, Xiaoyang Ding, Rong Li, Limei Zhao, and Chunbao Zhang. 2020. "Cytoplasm Types Affect DNA Methylation among Different Cytoplasmic Male Sterility Lines and Their Maintainer Line in Soybean (Glycine max L.)" Plants 9, no. 3: 385. https://doi.org/10.3390/plants9030385
APA StyleLin, C., Peng, B., Li, Y., Wang, P., Zhao, G., Ding, X., Li, R., Zhao, L., & Zhang, C. (2020). Cytoplasm Types Affect DNA Methylation among Different Cytoplasmic Male Sterility Lines and Their Maintainer Line in Soybean (Glycine max L.). Plants, 9(3), 385. https://doi.org/10.3390/plants9030385