Dissecting the Genetic Basis of Lateral and Central Spikelet Development and Grain Traits in Intermedium-Spike Barley (Hordeum vulgare Convar. Intermedium)

 , and
, and
Abstract
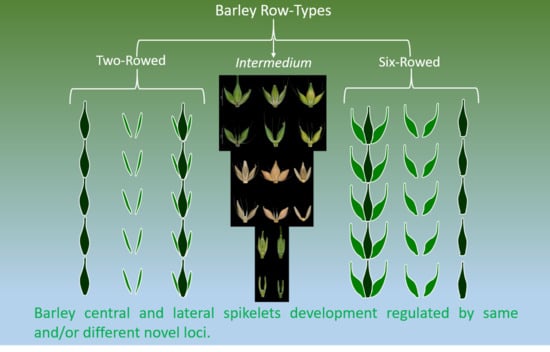
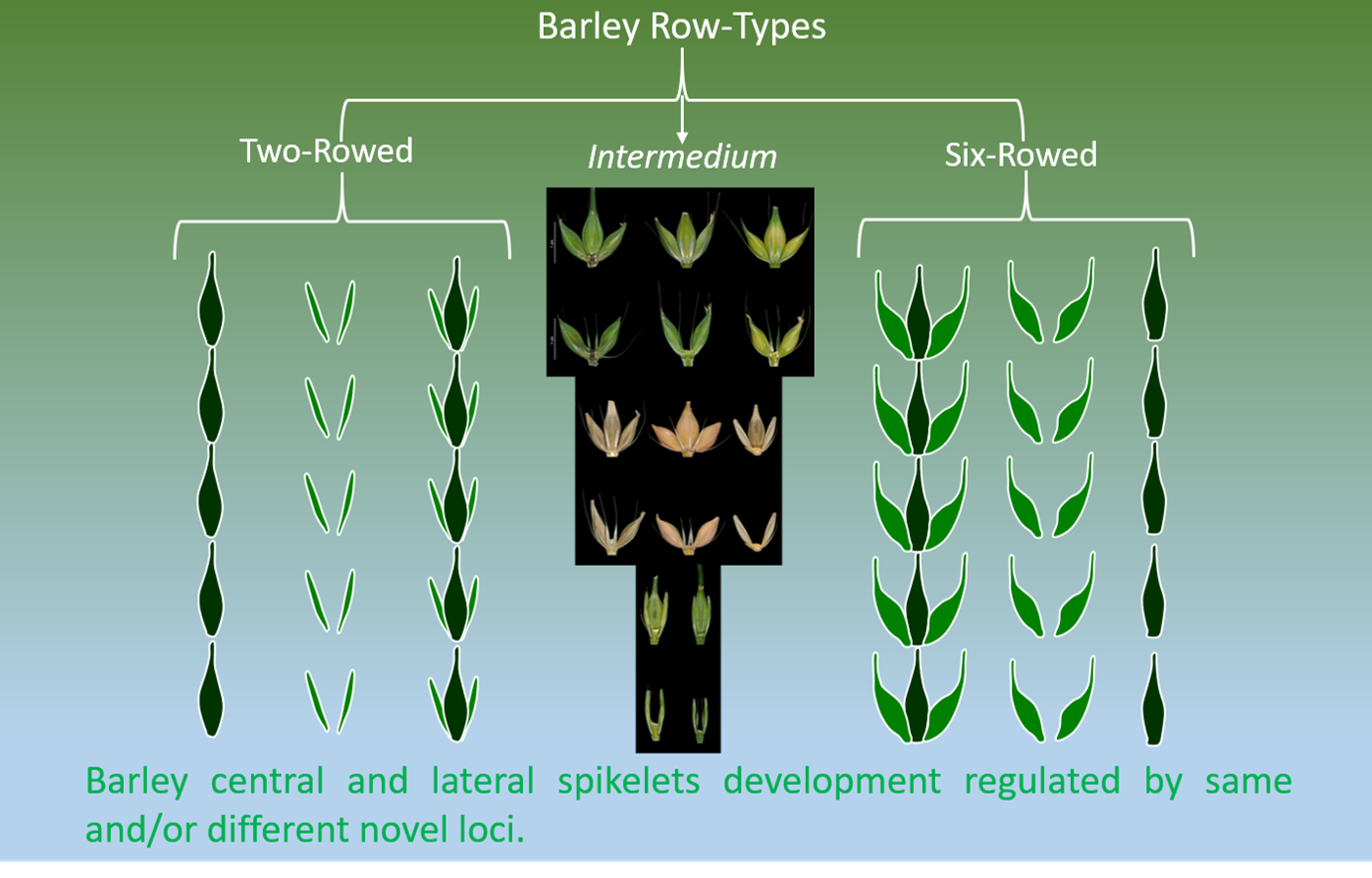
1. Introduction
2. Results
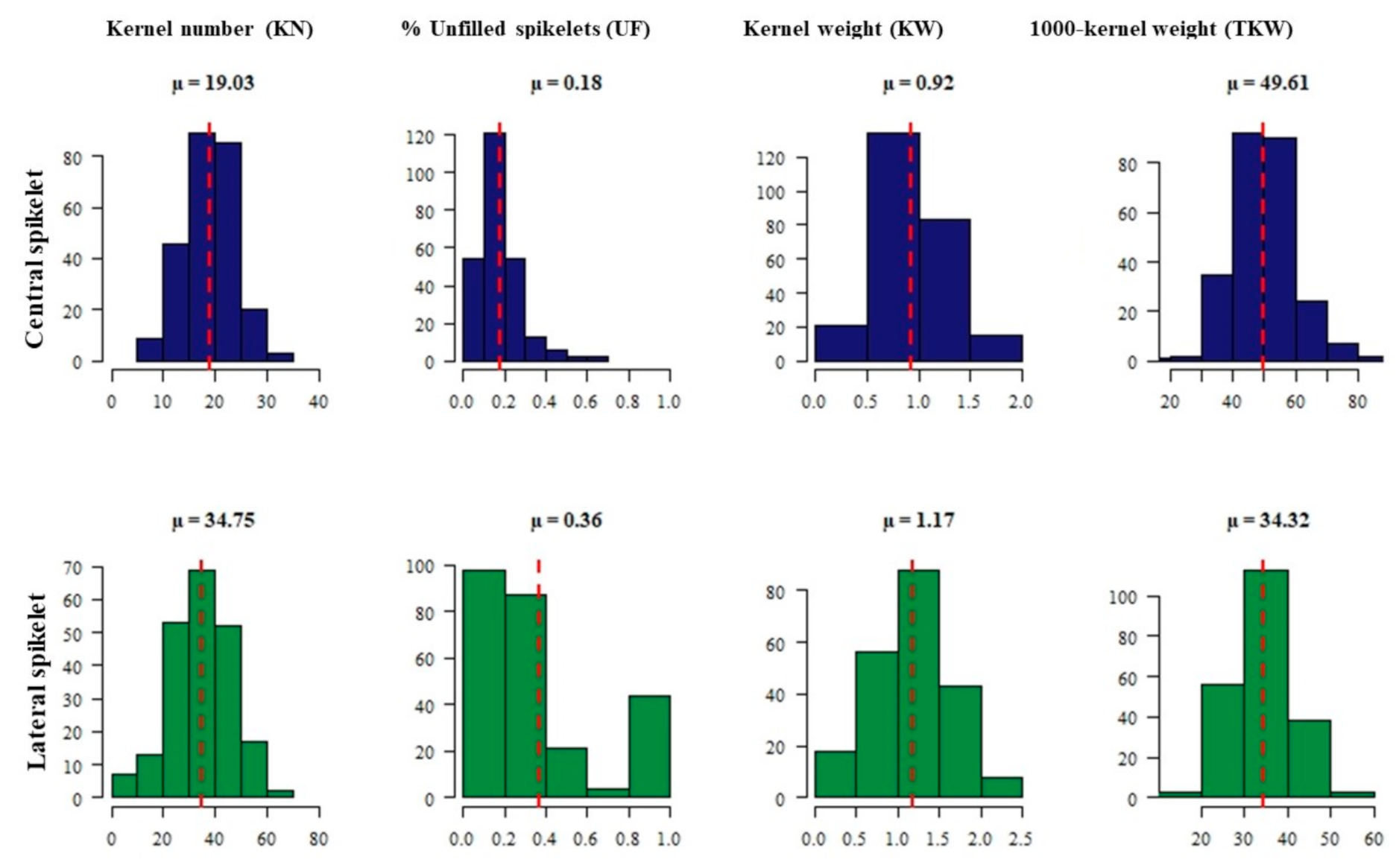
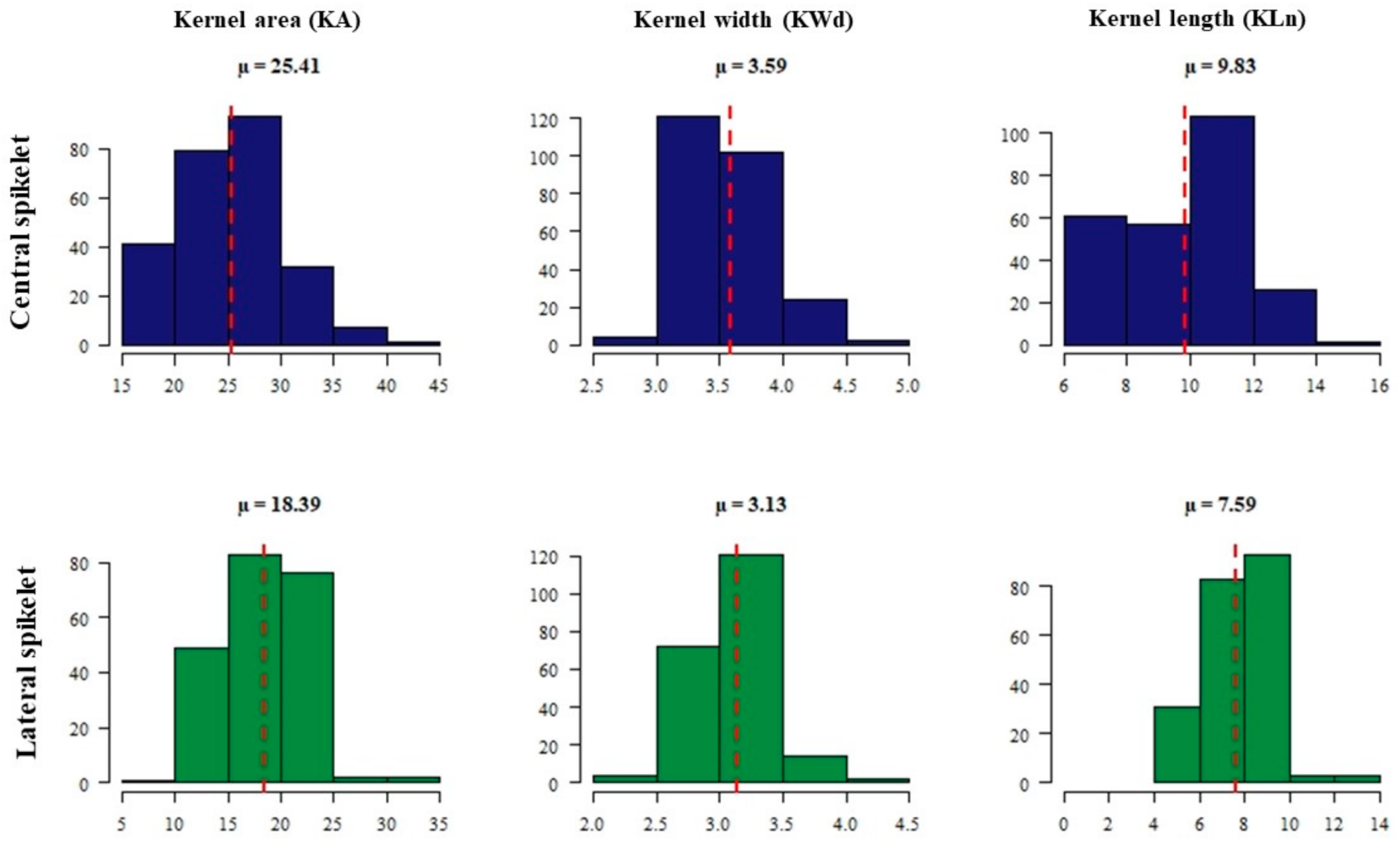
2.1. Phenotypic Data Analysis
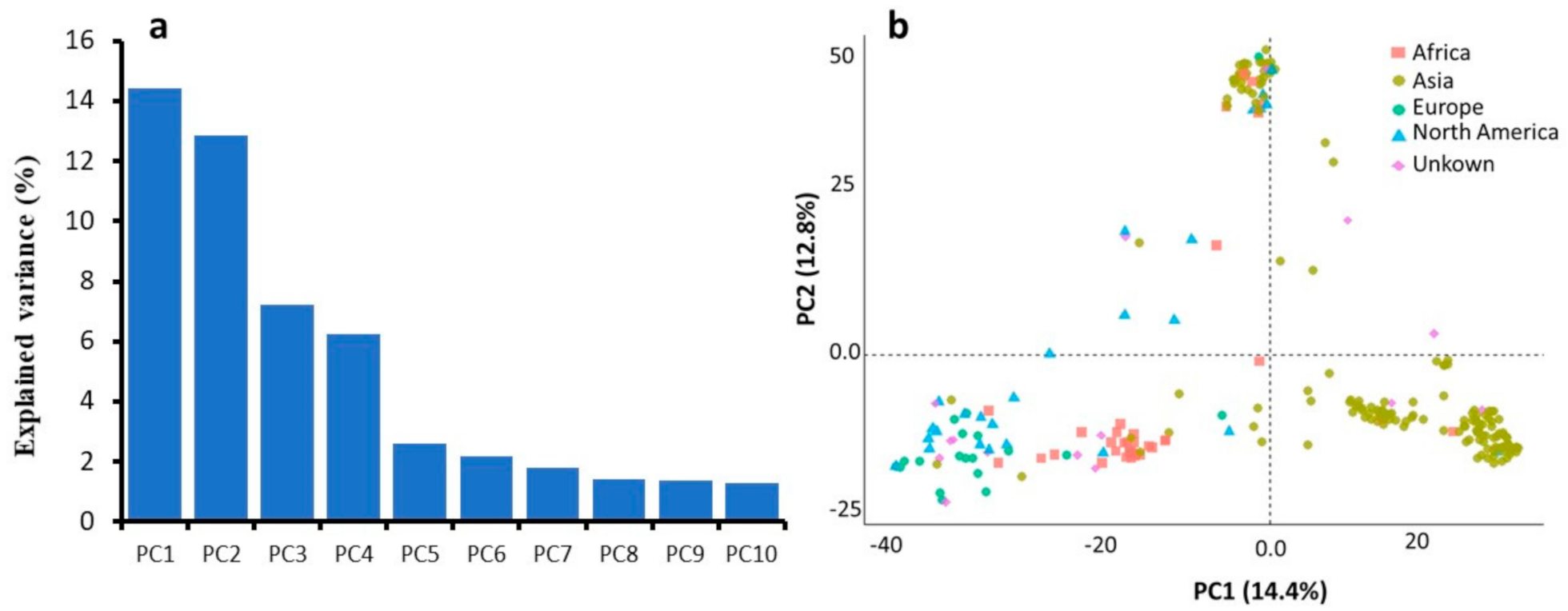
2.2. SNP Data and Population Structure Analysis
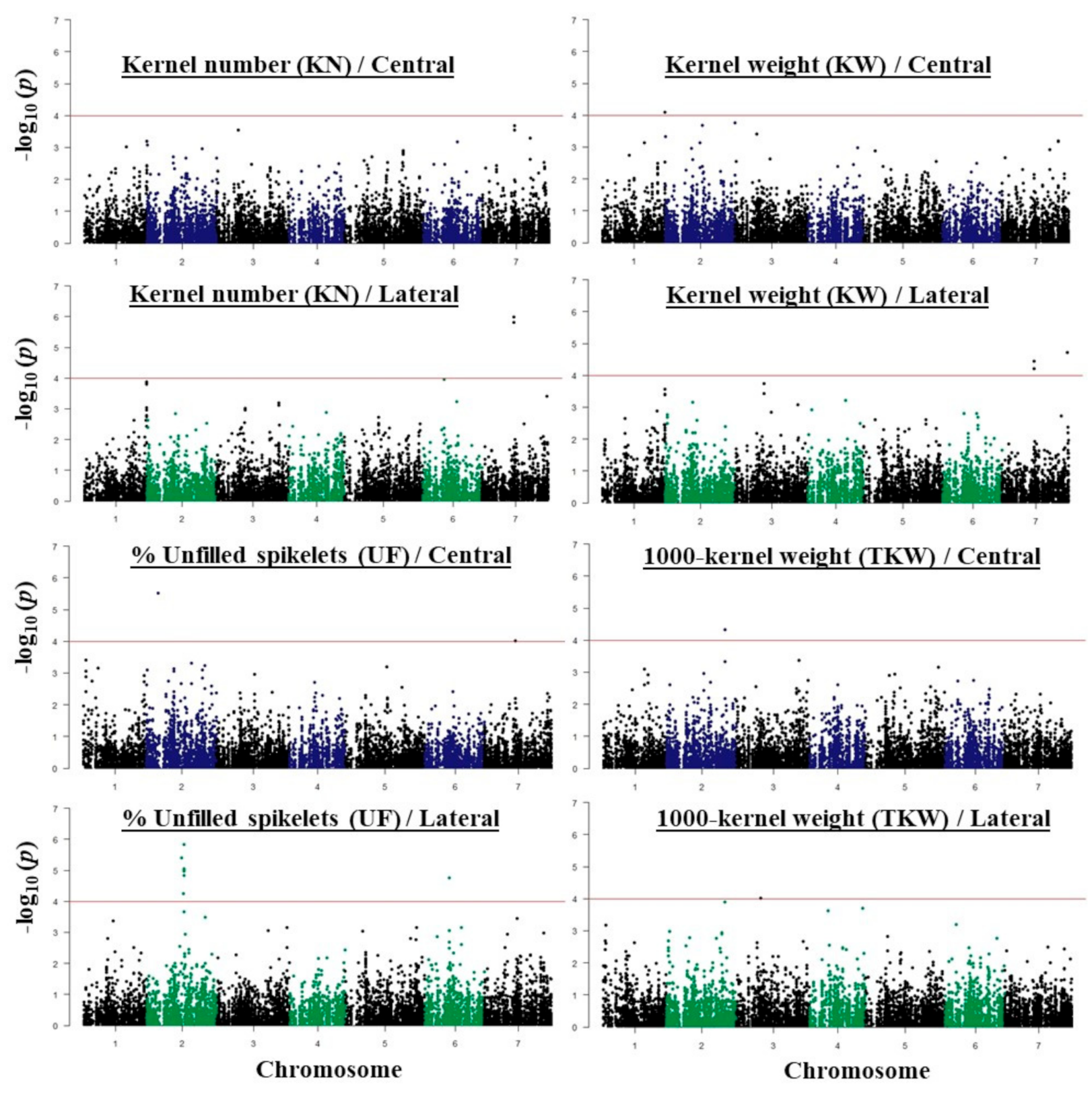
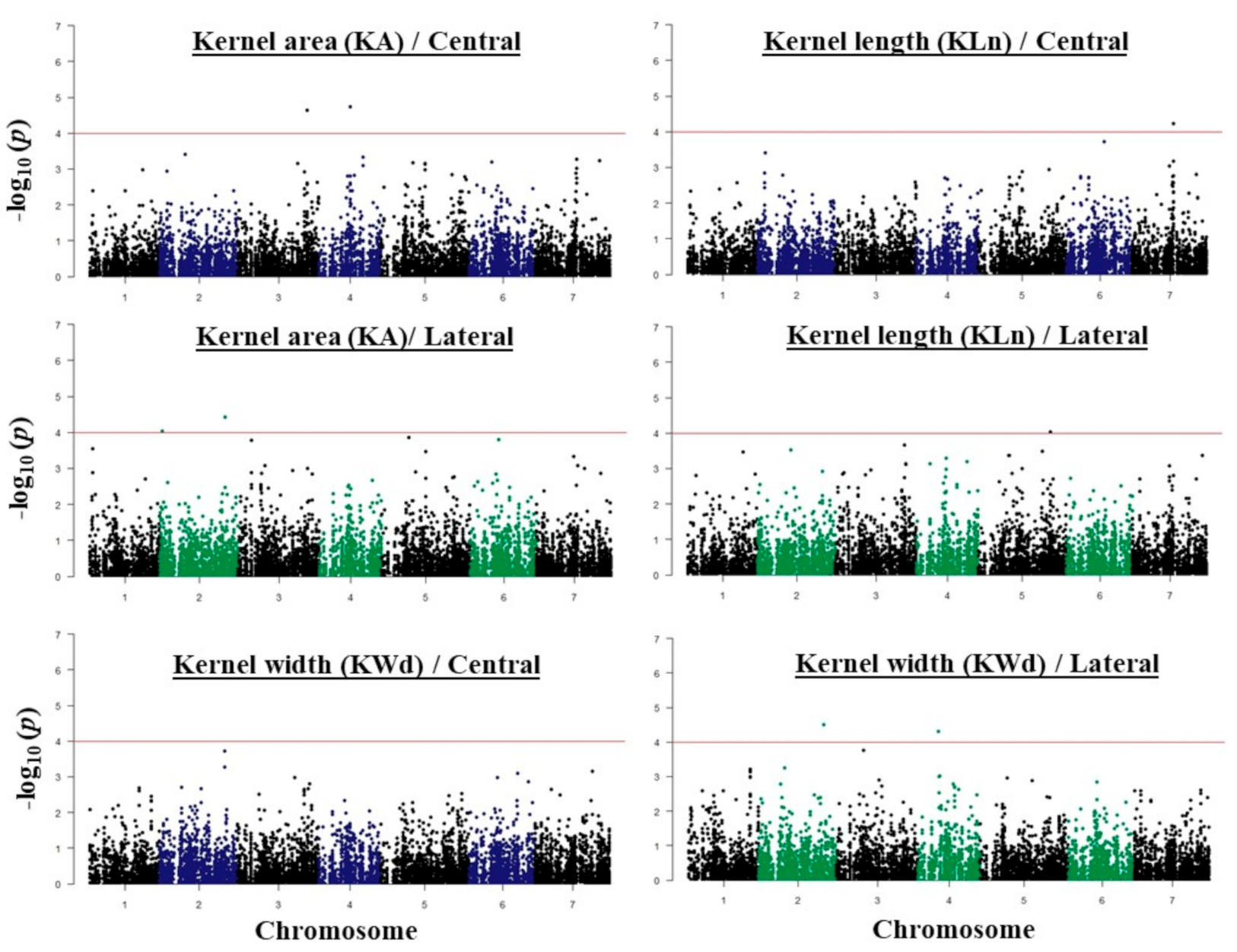
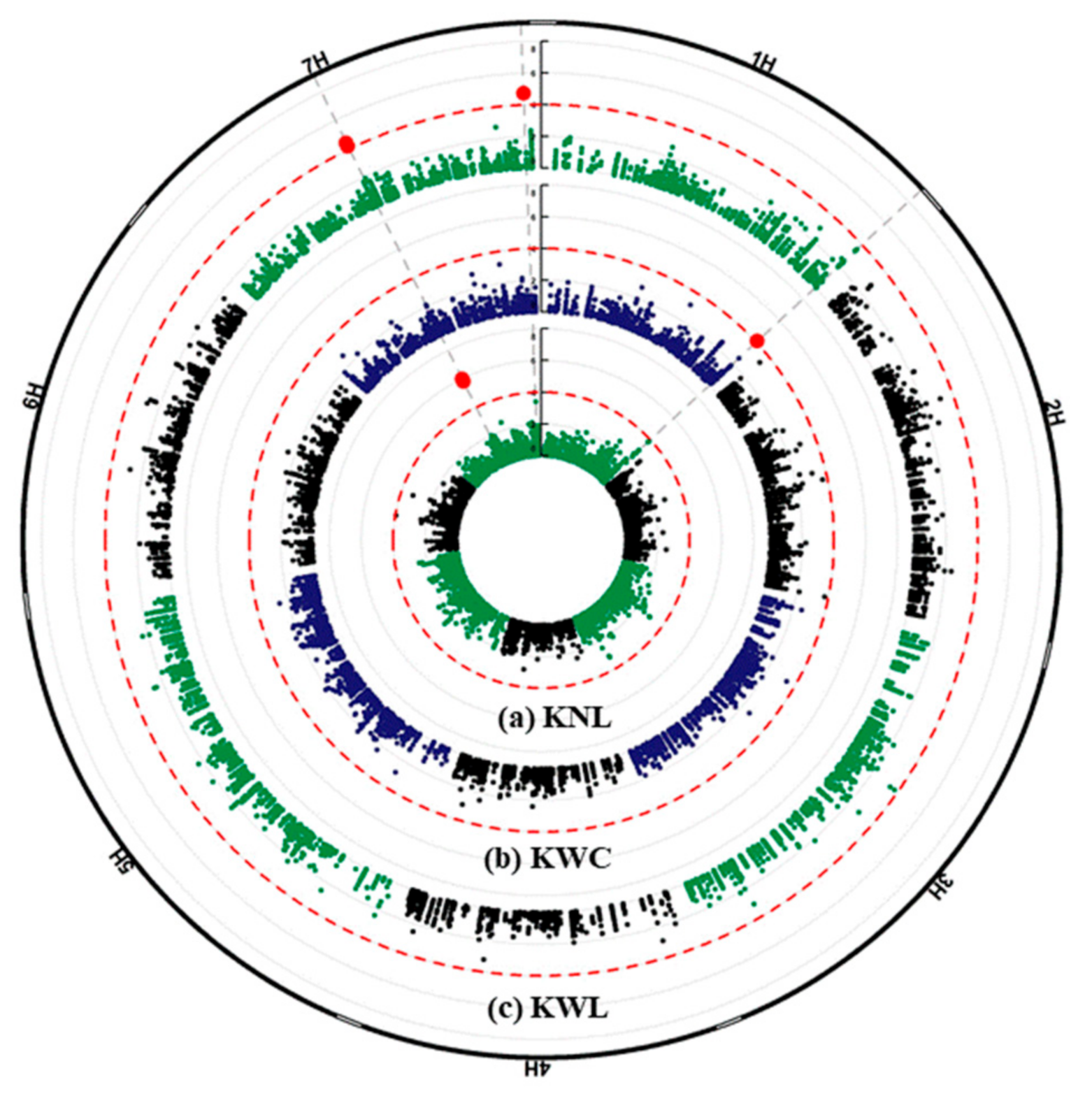
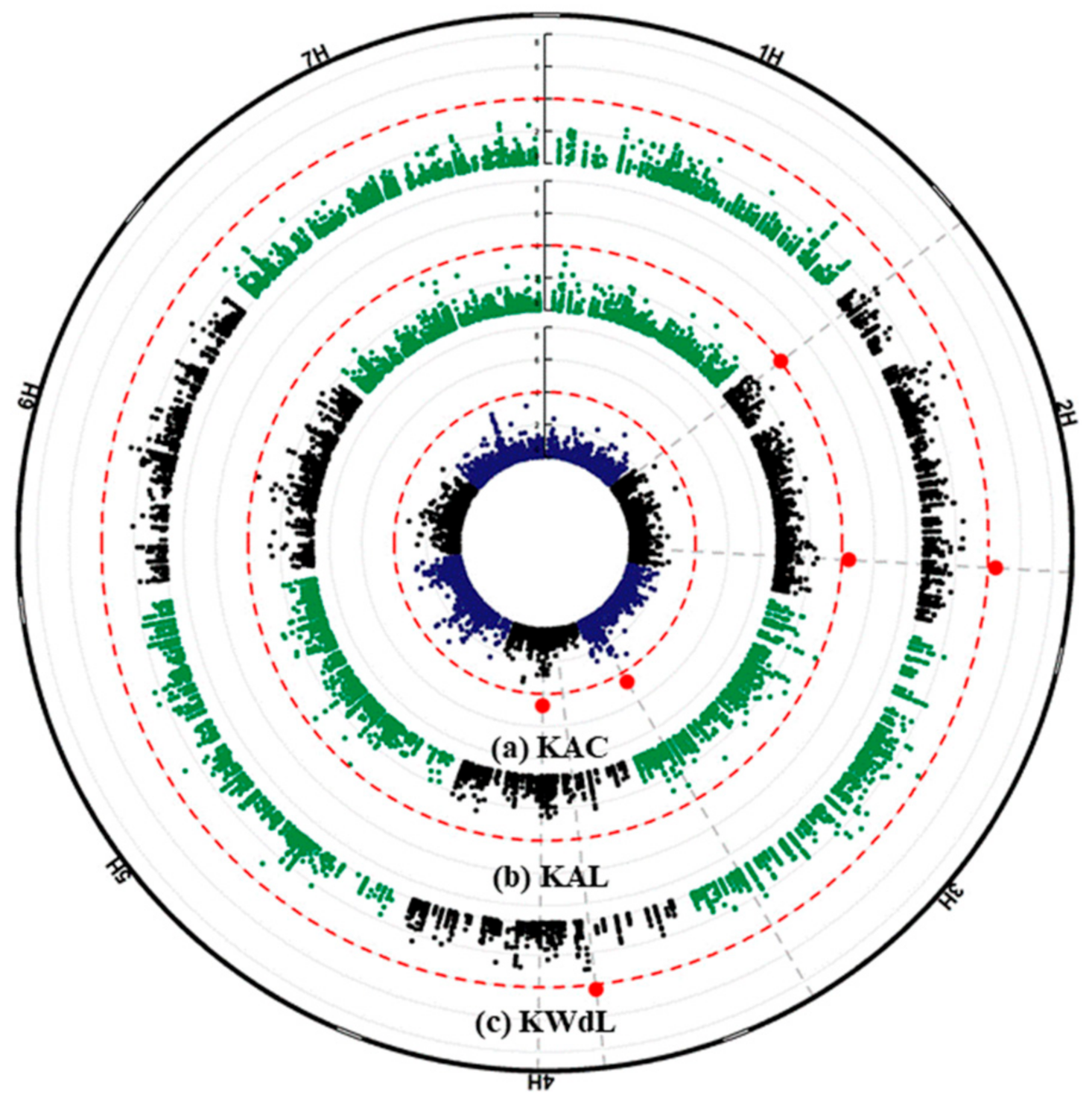
2.3. GWAS Analysis and Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Genetic Materials and Phenotyping
4.2. Phenotypic Data Analysis
4.3. GBS-SNP Genotyping
4.4. Population Structure and Kinship Analyses
4.5. Genome-Wide Association Mapping Analysis and Candidate Gene Identification
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAOSTAT. Food and Agriculture Organization of the United Nations (FAO); FAOSTAT Database; FAO: Rome, Italy, 2016. [Google Scholar]
- Koppolu, R.; Schnurbusch, T. Developmental pathways for shaping spike inflorescence architecture in barley and wheat. J. Integr. Plant Biol. 2019, 61, 278–295. [Google Scholar] [CrossRef]
- Abbo, S.; van-Oss, R.P.; Gopher, A.; Saranga, Y.; Ofner, I.; Peleg, Z. Plant domestication versus crop evolution: A conceptual framework for cereals and grain legumes. Trends Plant Sci. 2014, 19, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Schreiber, M.; Mascher, M. Domestication and crop evolution of wheat and barley: Genes, genomics, and future directions. J. Integr. Plant Biol. 2019, 61, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Knüpffer, H. Triticeae genetic resources in ex situ genebank collections. In Genetics and Genomics of the Triticeae, Plant Genetics and Genomics: Crops and Models 7; Muehlbauer, G.J., Feuillet, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 31–79. [Google Scholar]
- Allam, M.; Ordás, B.; Djemel, A.; Tracy, W.F.; Revilla, P. Linkage disequilibrium between fitness QTLs and the sugary1 allele of maize. Mol. Breed. 2019, 39, 3. [Google Scholar] [CrossRef]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Bayer, M. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Mansfeld, R. Das morphologische System der Saatgerste, Hordeum vulgare L. Theor. Appl. Genet. 1950, 20, 8–24. [Google Scholar] [CrossRef]
- Komatsuda, T.; Pourkheirandish, M.; He, C.; Azhaguvel, P.; Kanamori, H.; Perovic, D.; Lundqvist, U. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 2007, 104, 1424–1429. [Google Scholar] [CrossRef]
- Sakuma, S.; Pourkheirandish, M.; Hensel, G.; Kumlehn, J.; Stein, N.; Tagiri, A.; Yamaji, N.; Ma, J.F.; Sassa, H.; Koba, T.; et al. Divergence of expression pattern contributed to neofunctionalization of duplicated HD-Zip I transcription factor in barley. New Phytol. 2013, 197, 939–948. [Google Scholar] [CrossRef]
- Shuai, B.; Reynaga-Peña, C.; Springer, P. The lateral organ boundaries gene defines a novel, plant-specific gene family. Plant Physiol. 2002, 129, 747–761. [Google Scholar] [CrossRef]
- Ramsay, L.; Comadran, J.; Druka, A.; Marshall, D.F.; Thomas, W.T.; Macaulay, M.; Hayes, P.M. INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat. Genet. 2011, 43, 169–172. [Google Scholar] [CrossRef]
- Koppolu, R.; Anwar, N.; Sakuma, S.; Tagiri, A.; Lundqvist, U.; Pourkheirandish, M.; Rutten, T.; Seiler, C.; Himmelbach, A.; Ariyadasa, R.; et al. Six-rowed spike4 (Vrs4) controls spikelet determinacy and row-type in barley. Proc. Natl. Acad. Sci. USA 2013, 110, 13198–13203. [Google Scholar] [CrossRef] [PubMed]
- Youssef, H.M.; Eggert, K.; Koppolu, R.; Alqudah, A.M.; Poursarebani, N.; Fazeli, A.; Lundqvist, U. VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat. Genet. 2017, 49, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Bull, H.; Casao, M.C.; Zwirek, M.; Flavell, A.J.; Thomas, W.T.; Guo, W.; Druka, A. Barley SIX-ROWED SPIKE3 encodes a putative Jumonji C-type H3K9me2/me3 demethylase that represses lateral spikelet fertility. Nat. Commun. 2017, 8, 936. [Google Scholar] [CrossRef] [PubMed]
- van Esse, G.W.; Walla, A.; Finke, A.; Koornneef, M.; Pecinka, A.; von Korff, M. Six-Rowed Spike3 (VRS3) is a histone demethylase that controls lateral spikelet development in barley. Plant Physiol. 2017, 174, 2397–2408. [Google Scholar] [CrossRef]
- Pourkheirandish, M.; Komatsuda, T. The importance of barley genetics and domestication in a global perspective. Ann. Bot. 2007, 100, 999–1008. [Google Scholar] [CrossRef]
- Youssef, H.M.; Mascher, M.; Ayoub, M.A.; Stein, N.; Kilian, B.; Schnurbusch, T. Natural diversity of inflorescence architecture traces cryptic domestication genes in barley (Hordeum vulgare L.). Genet. Resour. Crop Evol. 2017, 64, 843–853. [Google Scholar] [CrossRef]
- Wenzl, P.; Raman, H.; Wang, J.; Zhou, M.; Huttner, E.; Kilian, A. A DArT platform for quantitative bulked segregant analysis. BMC Genom. 2007, 8, 196. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Nelson, M.N.; Aslam, M.N.; Rajasekaran, R.; Wratten, N.; Schondelmaier, J. Diversity Array Technology markers: Genetic diversity analyses and linkage map construction in rapeseed (Brassica napus L.). DNA Res. 2012, 19, 51–65. [Google Scholar] [CrossRef]
- Waugh, R.; Jannink, J.; Muehlbauer, G.J.; Ramsay, L. The emergence of whole genome association scans in barley. Curr. Opin. Plant Biol. 2009, 12, 218–222. [Google Scholar] [CrossRef]
- George, A.W.; Cavanagh, C. Genome-wide association mapping in plants. Theor. Appl. Genet. 2015, 128, 1163–1174. [Google Scholar] [CrossRef]
- Liu, H.J.; Yan, J. Crop genome-wide association study: A harvest of biological relevance. Plant J. 2019, 97, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.; Draba, V.; Jiang, Y.; Schnaithmann, F.; Sharma, R.; Schumann, E.; Pillen, K. Modelling the genetic architecture of flowering time control in barley through nested association mapping. BMC Genom. 2015, 16, 290. [Google Scholar] [CrossRef] [PubMed]
- Sannemann, W.; Huang, B.E.; Mathew, B.; Léon, J. Multi-parent advanced generation inter-cross in barley: High-resolution quantitative trait locus mapping for flowering time as a proof of concept. Mol. Breed. 2015, 35, 86. [Google Scholar] [CrossRef]
- Ayoub, M.; Symons, S.; Edney, M.; Mather, D. QTLs affecting kernel size and shape in a two-rowed by six-rowed barley cross. Theor. Appl. Genet. 2002, 105, 237–247. [Google Scholar] [CrossRef]
- Walker, C.K.; Ford, R.; Muñoz-Amatriaín, M.; Panozzo, J.F. The detection of QTLs in barley associated with endosperm hardness, grain density, grain size and malting quality using rapid phenotyping tools. Theor. Appl. Genet. 2013, 126, 2533–2551. [Google Scholar] [CrossRef]
- Zhou, Q.; Dong, Y.; Shi, Q.; Zhang, L.; Chen, H.; Hu, C.; Li, Y. Verification and fine mapping of qGW1.05, a major QTL for grain weight in maize (Zea mays L.). Mol. Genet. Genom. 2017, 292, 871–881. [Google Scholar] [CrossRef]
- Xu, X.; Sharma, R.; Tondelli, A.; Russell, J.; Comadran, J.; Schnaithmann, F.; Flavell, A.J. Genome-wide association analysis of grain yield-associated traits in a pan-European barley cultivar collection. Plant Genome 2018, 11, 170073. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, G.; Ren, X.; Du, B.; Cheng, Y.; Wang, Y.; Sun, D. Dissecting the genetic basis of grain size and weight in barley (Hordeum vulgare L.) by QTL and comparative genetic analyses. Front. Plant Sci. 2019, 10, 469. [Google Scholar] [CrossRef]
- Comadran, J.; Russell, J.R.; Booth, A.; Pswarayi, A.; Ceccarelli, S.; Grando, S.; Benbelkacem, A. Mixed model association scans of multi-environmental trial data reveal major loci controlling yield and yield related traits in Hordeum vulgare in Mediterranean environments. Theor. Appl. Genet. 2011, 122, 1363–1373. [Google Scholar] [CrossRef]
- Kalladan, R.; Worch, S.; Rolletschek, H.; Harshavardhan, V.T.; Kuntze, L.; Seiler, C.; Röder, M.S. Identification of quantitative trait loci contributing to yield and seed quality parameters under terminal drought in barley advanced backcross lines. Mol. Breed. 2013, 32, 71–90. [Google Scholar] [CrossRef]
- Maurer, A.; Draba, V.; Pillen, K. Genomic dissection of plant development and its impact on thousand grain weight in barley through nested association mapping. J. Exp. Bot. 2016, 67, 2507–2518. [Google Scholar] [CrossRef] [PubMed]
- Sameri, M.; Komatsuda, T. Localization of quantitative trait loci for yield components in a cross oriental × occidental barley cultivar (Hordeum vulgare L.). Jpn. Agric. Res. Q. 2007, 41, 195–199. [Google Scholar] [CrossRef][Green Version]
- Cu, S.T.; March, T.J.; Stewart, S.; Degner, S.; Coventry, S.; Box, A.; Eglinton, J. Genetic analysis of grain and malt quality in an elite barley population. Mol. Breed. 2016, 36, 129. [Google Scholar] [CrossRef]
- Zwirek, M.; Waugh, R.; McKim, S.M. Interaction between row-type genes in barley controls meristem determinacy and reveals novel routes to improved grain. New Phytol. 2019, 221, 1950–1965. [Google Scholar] [CrossRef]
- Tsuda, K.; Tsuji, T.; Hirose, S.; Yamazaki, K. Three Arabidopsis MBF1 homologs with distinct expression profiles play roles as transcriptional co-activators. Plant Cell Physiol. 2004, 45, 225–231. [Google Scholar] [CrossRef]
- Sakuma, S.; Lundqvist, U.; Kakei, Y.; Thirulogachandar, V.; Suzuki, T.; Hori, K.; Shimada, Y. Extreme suppression of lateral floret development by a single amino acid change in the VRS1 transcription factor. Plant Physiol. 2017, 175, 1720–1731. [Google Scholar] [CrossRef]
- Huang, J.; Pang, C.; Fan, S.; Song, M.; Yu, J.; Wei, H.; Yu, S. Genome-wide analysis of the family 1 glycosyltransferases in cotton. Mol. Genet. Genom. 2015, 290, 1805–1818. [Google Scholar] [CrossRef]
- Xing, L.P.; He, L.Q.; Xiao, J.; Chen, Q.G.; Li, M.H.; Shang, Y.; Wang, X.E. An UDP-glucosyltransferase gene from barley confers disease resistance to Fusarium head blight. Plant Mol. Biol. Rep. 2017, 35, 224–236. [Google Scholar] [CrossRef]
- Boden, S.A.; Weiss, D.; Ross, J.J.; Davies, N.W.; Trevaskis, B.; Chandler, P.M.; Swain, S.M. EARLY FLOWERING 3 regulates flowering in spring barley by mediating gibberellin production and FLOWERING LOCUS T expression. Plant Cell 2014, 26, 1557–1569. [Google Scholar] [CrossRef]
- Colmsee, C.; Beier, S.; Himmelbach, A.; Schmutzer, T.; Stein, N.; Scholz, U.; Mascher, M. BARLEX—The Barley Draft Genome Explorer. Mol. Plant 2015, 964–966. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Mascher, M.; Wu, S.; Amand, P.S.; Stein, N.; Poland, J. Application of genotyping-by-sequencing on semiconductor sequencing platforms: A comparison of genetic and reference-based marker ordering in barley. PLoS ONE 2013, 8, e76925. [Google Scholar] [CrossRef] [PubMed]
- Mascher, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multi-locus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Earl, D.A.; von Holdt, B.M. Structure harvester: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. J. Bioinform. 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; Kresovich, S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and Manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | SNP ID | Chr | SNP Physical Position | Ref Allele | Alt Allele | Gene | HC/LC | Annotation |
|---|---|---|---|---|---|---|---|---|
| Kernel number “KN” | m1_556903593 | 1H | 556903593 | T | C | HORVU1Hr1G094980 | HC_G | Early flowering 3 (ELF3) |
| m7_168762979 | 7H | 168762979 | G | T | HORVU7Hr1G048880 | HC_G | Sec14p-like phosphatidylinositol transfer family protein | |
| m7_168763009 | 7H | 168763009 | C | T | HORVU7Hr1G048880 | HC_G | Sec14p-like phosphatidylinositol transfer family protein | |
| % Unfilled spikelets “UF” | m2_35441939 | 2H | 35441939 | C | T | HORVU2Hr1G001640 | HC_G | Mediator of RNA polymerase II transcription subunit 15a |
| m7_109046266 | 7H | 109046266 | C | T | HORVU7Hr1G040320 | LC_u | undescribed protein | |
| Kernel weight “KW” | m1_556903593 | 1H | 556903593 | T | C | HORVU1Hr1G094980 | HC_G | Early flowering 3 |
| m2_647258179 | 2H | 647258179 | A | G | HORVU2Hr1G091170 | HC_G | Expansin B3 | |
| m2_653986096 | 653986096 | C | A | HORVU2Hr1G092530 | HC_G | Aldehyde dehydrogenase family 3 member F1 | ||
| Thousand kernel weight “TKW” | m2_727927744 | 2H | 727927744 | T | C | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein |
| m2_727927750 | 2H | 727927750 | G | T | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m2_727927781 | 2H | 727927781 | G | A | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m3_661788536 | 3H | 661788539 | A | C | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| m3_661788539 | 3H | 661788539 | T | G | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| m3_661788540 | 3H | 661788539 | A | G | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| m3_661788542 | 3H | 661788539 | A | T | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| Kernel area “KA” | m3_661788536 | 3H | 661788539 | A | C | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein |
| m3_661788539 | 3H | 661788539 | T | G | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| m3_661788540 | 3H | 661788539 | A | G | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| m3_661788542 | 3H | 661788539 | A | T | HORVU3Hr1G099530 | HC_G | UDP-Glycosyltransferase superfamily protein | |
| m4_557589531 | 4H | 557589531 | C | T | HORVU4Hr1G067140 | HC_G | Protein kinase superfamily protein | |
| m7_548419008 | 7H | 548419008 | G | T | HORVU7Hr1G090180 | LC_u | undescribed protein | |
| Kernel width “KWd” | m2_727927744 | 2H | 727927744 | T | C | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein |
| m2_727927750 | 2H | 727927750 | G | T | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m2_727927781 | 2H | 727927781 | G | A | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| Kernel Length “KLn” | m7_548419008 | 7H | 548419008 | G | T | HORVU7Hr1G090180 | LC_u | undescribed protein |
| Traits | SNP ID | Chr | SNP Physical Position | Ref Allele | Alt Allele | Gene | HC/LC | Annotation |
|---|---|---|---|---|---|---|---|---|
| Kernel number “KN” | m7_168762979 | 7H | 168762979 | G | T | HORVU7Hr1G048880 | HC_G | Sec14p-like phosphatidylinositol transfer family protein |
| m7_168763009 | 7H | 168763009 | C | T | HORVU7Hr1G048880 | HC_G | Sec14p-like phosphatidylinositol transfer family protein | |
| m7_651215909 | 7H | 651215909 | C | T | HORVU7Hr1G120030 | HC_G | Delta(24)-sterol reductase | |
| % Unfilled spikelets “UF” | m2_647258179 | 2H | 647258179 | A | G | HORVU2Hr1G091170 | HC_G | Expansin B3 |
| m2_649489024 | 2H | 649489024 | G | A | HORVU2Hr1G091760 | LC_TE | Retrotransposon protein, putative, unclassified | |
| m2_649557855 | 2H | 649557855 | T | C | HORVU2Hr1G091830 | HC_G | undescribed protein | |
| m2_651372029 | 2H | 651372029 | G | A | HORVU2Hr1G092180 | HC_G | Elongation factor G | |
| m2_653986096 | 2H | 653986096 | C | A | HORVU2Hr1G092530 | HC_G | Aldehyde dehydrogenase family 3 member F1 | |
| m2_654165703 | 2H | 654165703 | A | G | HORVU2Hr1G092600 | HC_G | multiprotein bridging factor 1A | |
| m2_654165718 | 2H | 654165718 | C | G | HORVU2Hr1G092600 | HC_G | multiprotein bridging factor 1A | |
| m2_654165739 | 2H | 654165739 | C | A | HORVU2Hr1G092600 | HC_G | multiprotein bridging factor 1A | |
| m6_115087828 | 6H | 115087828 | G | T | HORVU6Hr1G028720 | HC_G | V-type ATP synthase subunit D | |
| Kernel weight “KW” | m7_168762979 | 7H | 168762979 | G | T | HORVU7Hr1G048880 | HC_G | Sec14p-like phosphatidylinositol transfer family protein |
| m7_168763009 | 7H | 168763009 | C | T | HORVU7Hr1G048880 | HC_G | Sec14p-like phosphatidylinositol transfer family protein | |
| m7_651215909 | 7H | 651215909 | C | T | HORVU7Hr1G120030 | HC_G | Delta(24)-sterol reductase | |
| Thousand kernel weight “TKW” | m2_727927744 | 2H | 727927744 | T | C | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein |
| m2_727927750 | 2H | 727927750 | G | T | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m3_208358229 | 3H | 208358229 | C | A | HORVU3Hr1G036930 | HC_G | Mediator of RNA polymerase II transcription subunit 27 | |
| m4_26351423 | 4H | 26351423 | G | A | HORVU4Hr1G009300 | HC_G | HXXXD-type acyl-transferase family protein | |
| Kernel area “KA” | m2_8906731 | 2H | 8906731 | T | G | HORVU2Hr1G003720 | HC_G | GDSL esterase/lipase |
| m2_727927744 | 2H | 727927744 | T | C | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m2_727927750 | 2H | 727927750 | G | T | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m3_208358229 | 3H | 208358229 | C | A | HORVU3Hr1G036930 | HC_G | Mediator of RNA polymerase II transcription subunit 27 | |
| m7_548419008 | 7H | 548419008 | G | T | HORVU7Hr1G090180 | LC_u | undescribed protein | |
| Kernel width “KWd” | m2_727927744 | 2H | 727927744 | T | C | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein |
| m2_727927750 | 2H | 727927750 | G | T | HORVU2Hr1G113260 | HC_G | N-terminal nucleophile aminohydrolases (Ntn hydrolases) superfamily protein | |
| m3_208358229 | 3H | 208358229 | C | A | HORVU3Hr1G036930 | HC_G | Mediator of RNA polymerase II transcription subunit 27 | |
| m4_26351423 | 4H | 26351423 | G | A | HORVU4Hr1G009300 | HC_G | HXXXD-type acyl-transferase family protein | |
| Kernel Length “KLn” | m5_619972700 | 5H | 619972700 | A | G | HORVU5Hr1G104750 | HC_G | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein|none |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssef, H.M.; Allam, M.; Boussora, F.; Himmelbach, A.; Milner, S.G.; Mascher, M.; Schnurbusch, T. Dissecting the Genetic Basis of Lateral and Central Spikelet Development and Grain Traits in Intermedium-Spike Barley (Hordeum vulgare Convar. Intermedium). Plants 2020, 9, 1655. https://doi.org/10.3390/plants9121655
Youssef HM, Allam M, Boussora F, Himmelbach A, Milner SG, Mascher M, Schnurbusch T. Dissecting the Genetic Basis of Lateral and Central Spikelet Development and Grain Traits in Intermedium-Spike Barley (Hordeum vulgare Convar. Intermedium). Plants. 2020; 9(12):1655. https://doi.org/10.3390/plants9121655
Chicago/Turabian StyleYoussef, Helmy M., Mohamed Allam, Faiza Boussora, Axel Himmelbach, Sara G. Milner, Martin Mascher, and Thorsten Schnurbusch. 2020. "Dissecting the Genetic Basis of Lateral and Central Spikelet Development and Grain Traits in Intermedium-Spike Barley (Hordeum vulgare Convar. Intermedium)" Plants 9, no. 12: 1655. https://doi.org/10.3390/plants9121655
APA StyleYoussef, H. M., Allam, M., Boussora, F., Himmelbach, A., Milner, S. G., Mascher, M., & Schnurbusch, T. (2020). Dissecting the Genetic Basis of Lateral and Central Spikelet Development and Grain Traits in Intermedium-Spike Barley (Hordeum vulgare Convar. Intermedium). Plants, 9(12), 1655. https://doi.org/10.3390/plants9121655