Seed Dormancy Involves a Transcriptional Program That Supports Early Plastid Functionality during Imbibition

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Germination Tests
2.2. General Assessment of the RNA-Seq Results
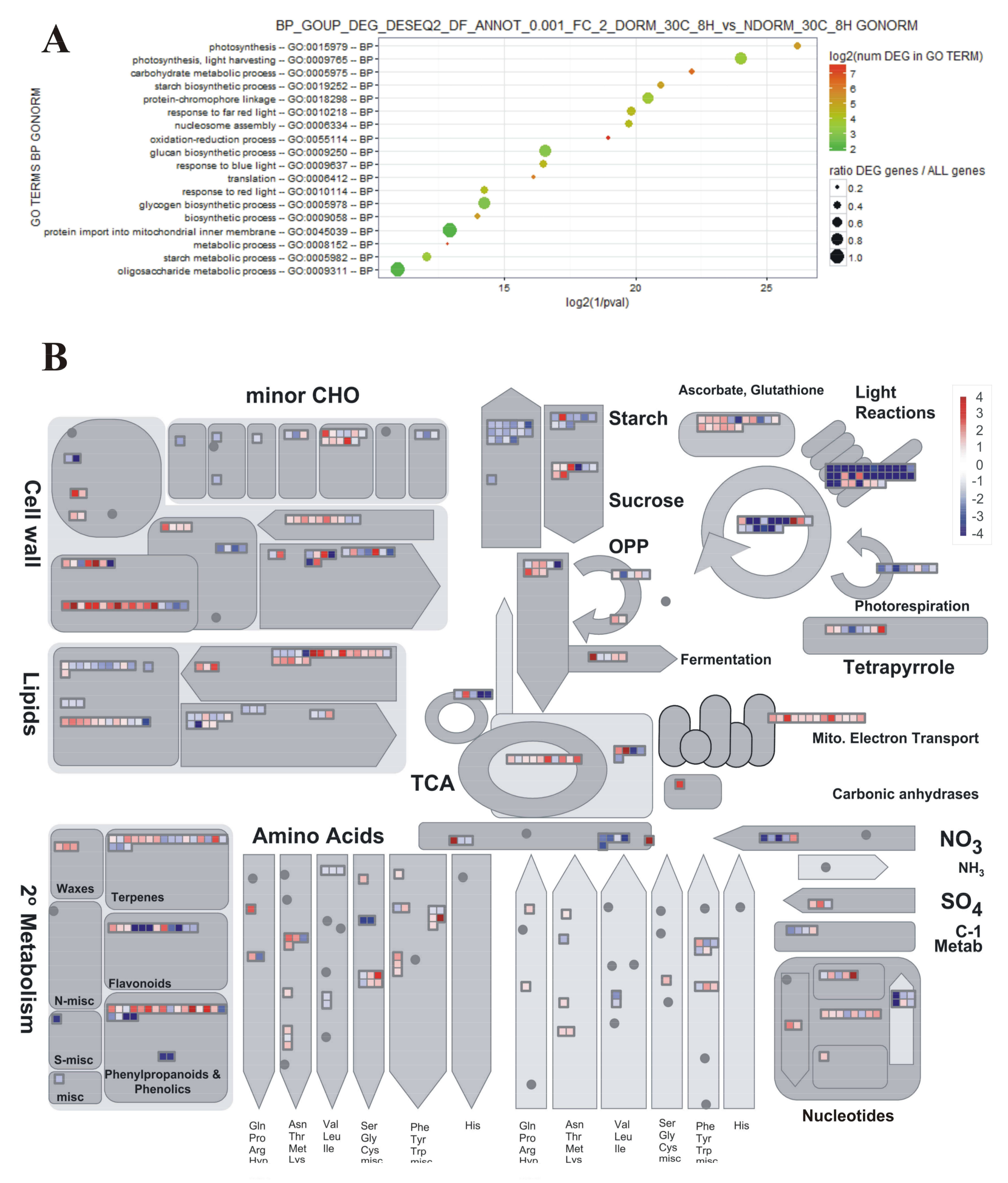
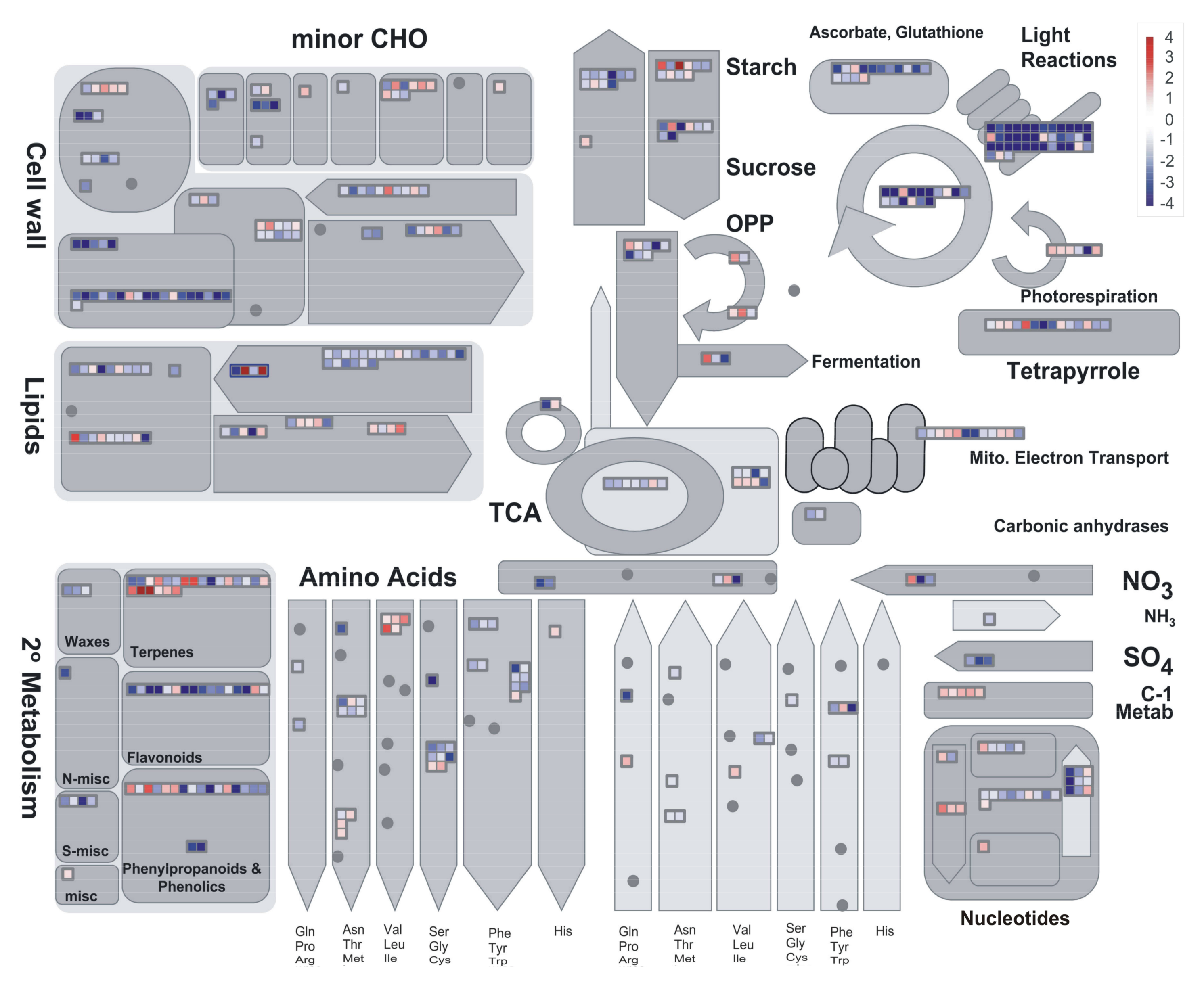
2.3. Preliminary Assessment of Expression Profiles with PageMan
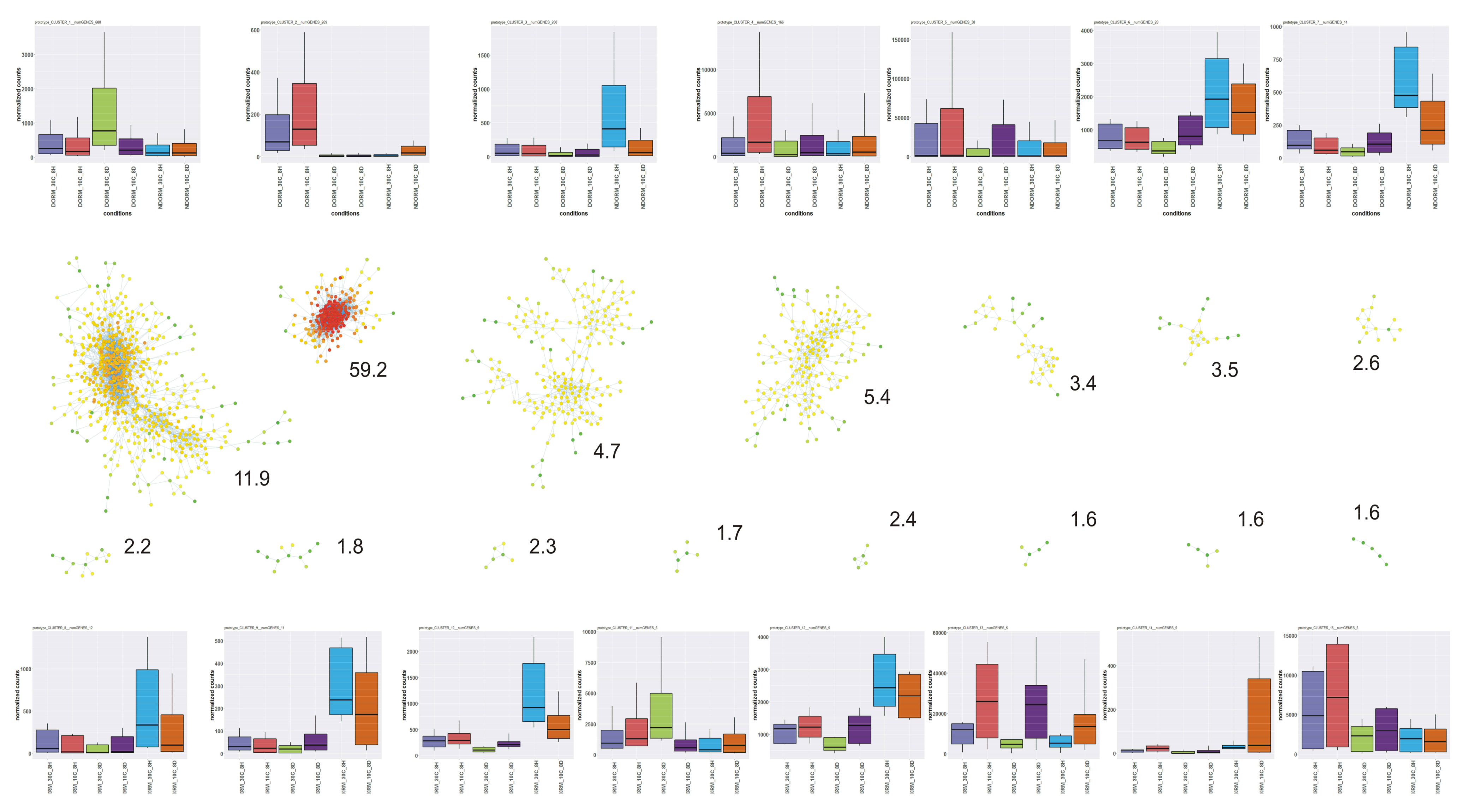
2.4. DEGs Classification and Analysis
2.5. Long Non-Coding RNAs
3. Discussion
3.1. The Impairing Effect of Dry-Afterripening
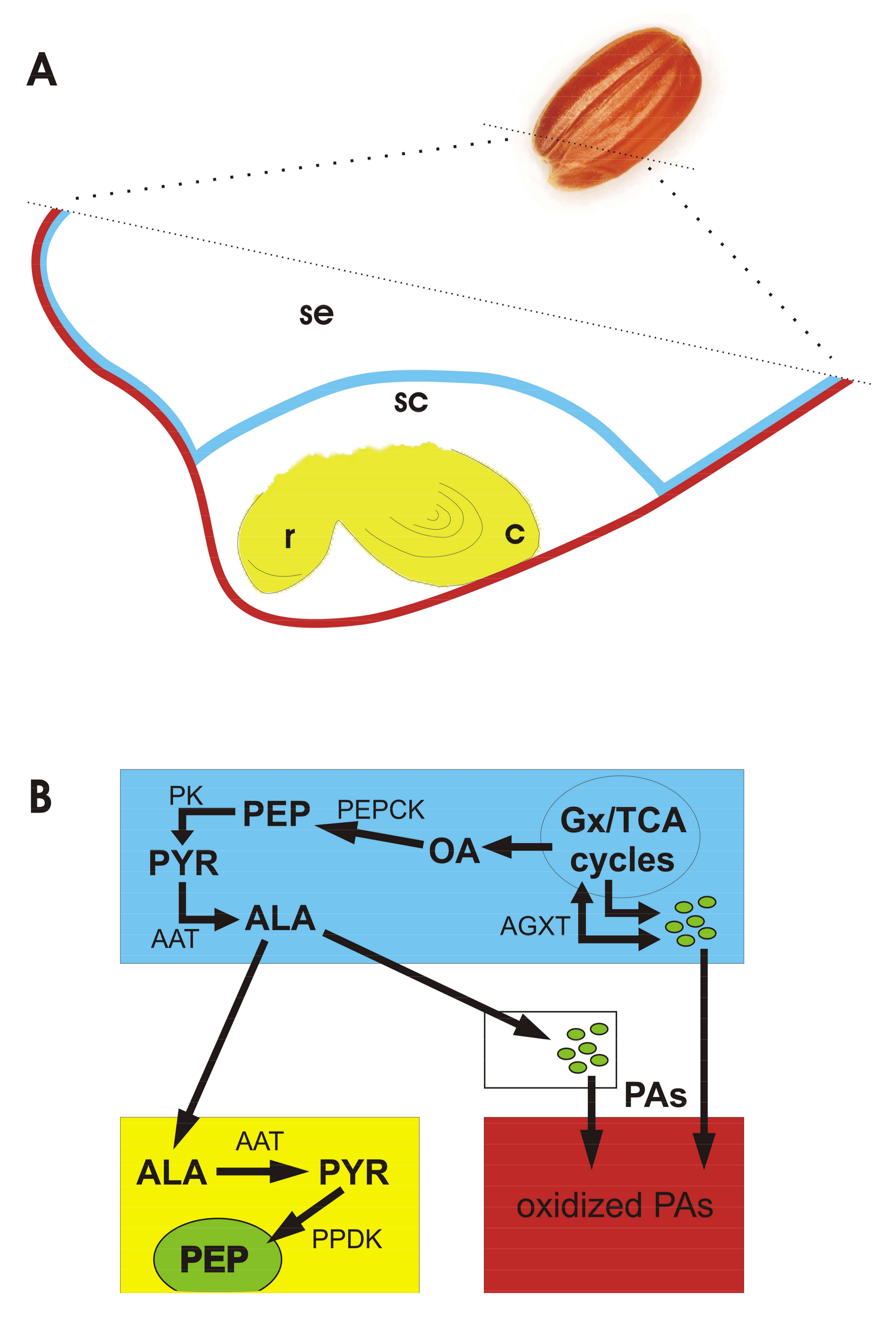
3.2. Nitrogen Metabolism
3.3. Carbon Metabolism
3.4. Phosphoenolpyruvate Carboxykinase (PEPCK)
3.5. Alanine Nutritional Shuttle
3.6. Further Sugar Metabolism Features
3.7. Cell Wall Modifying Enzymes
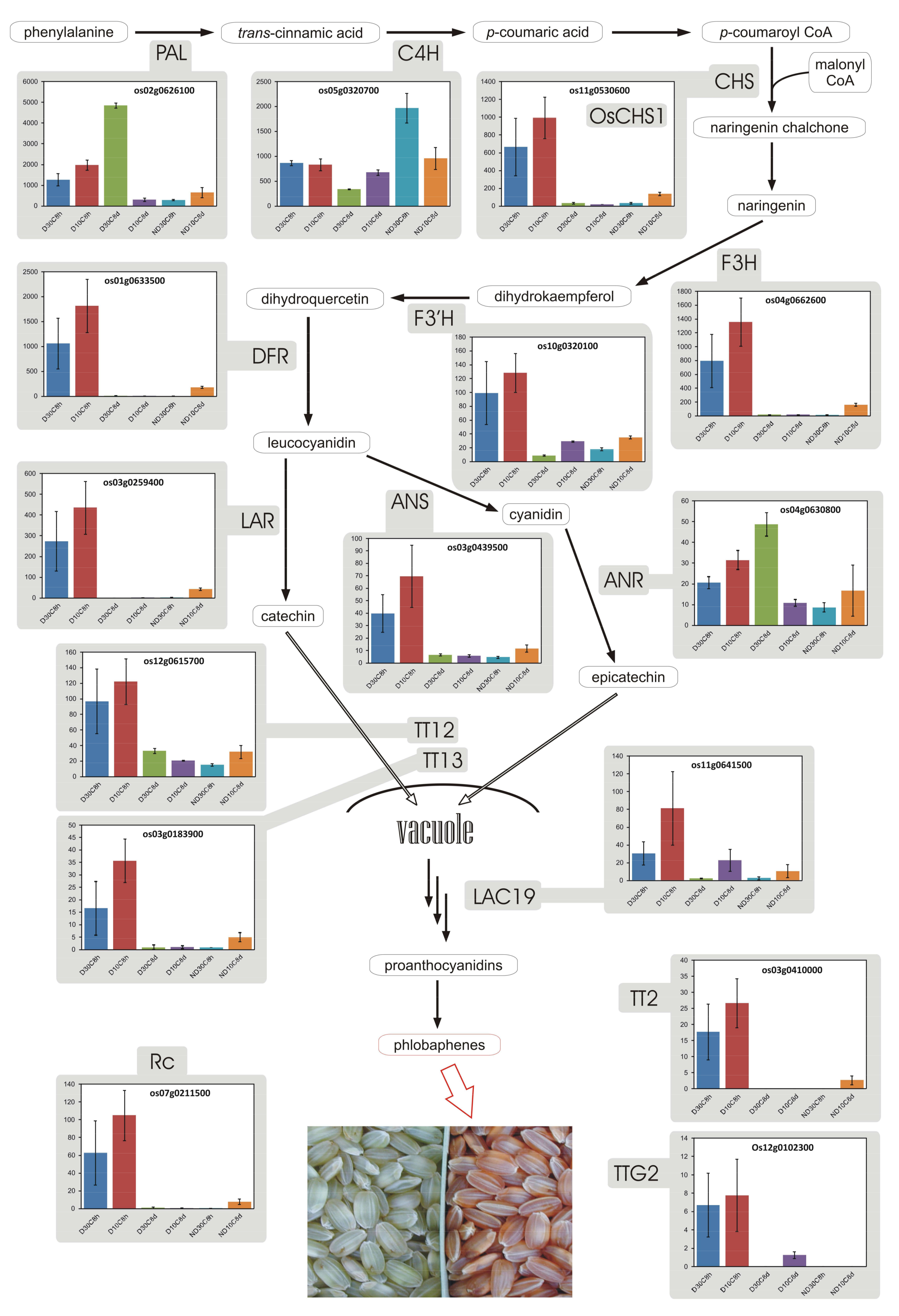
3.8. Proanthocyanidins and Phlobaphenes
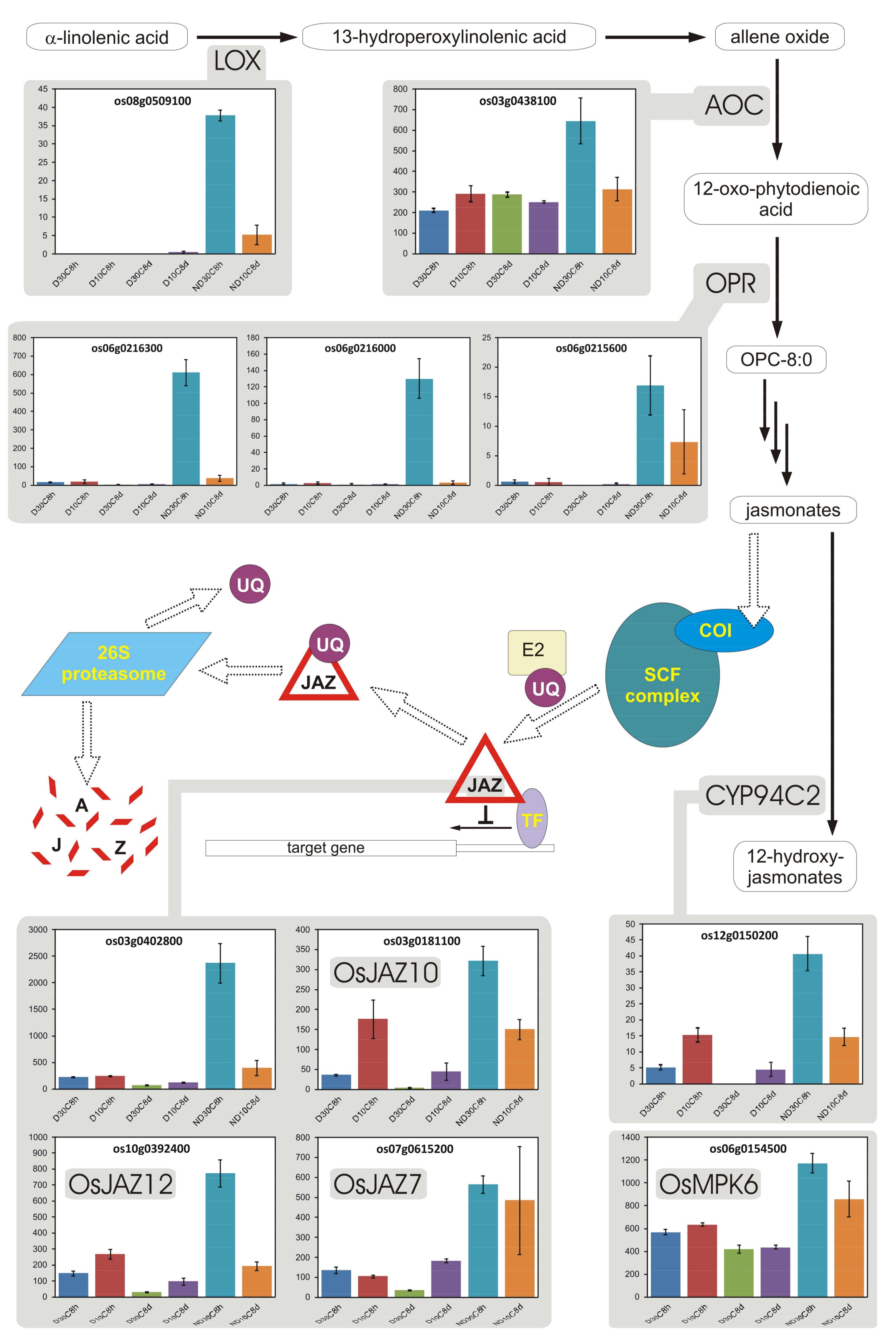
3.9. Jasmonates
3.10. Auxin
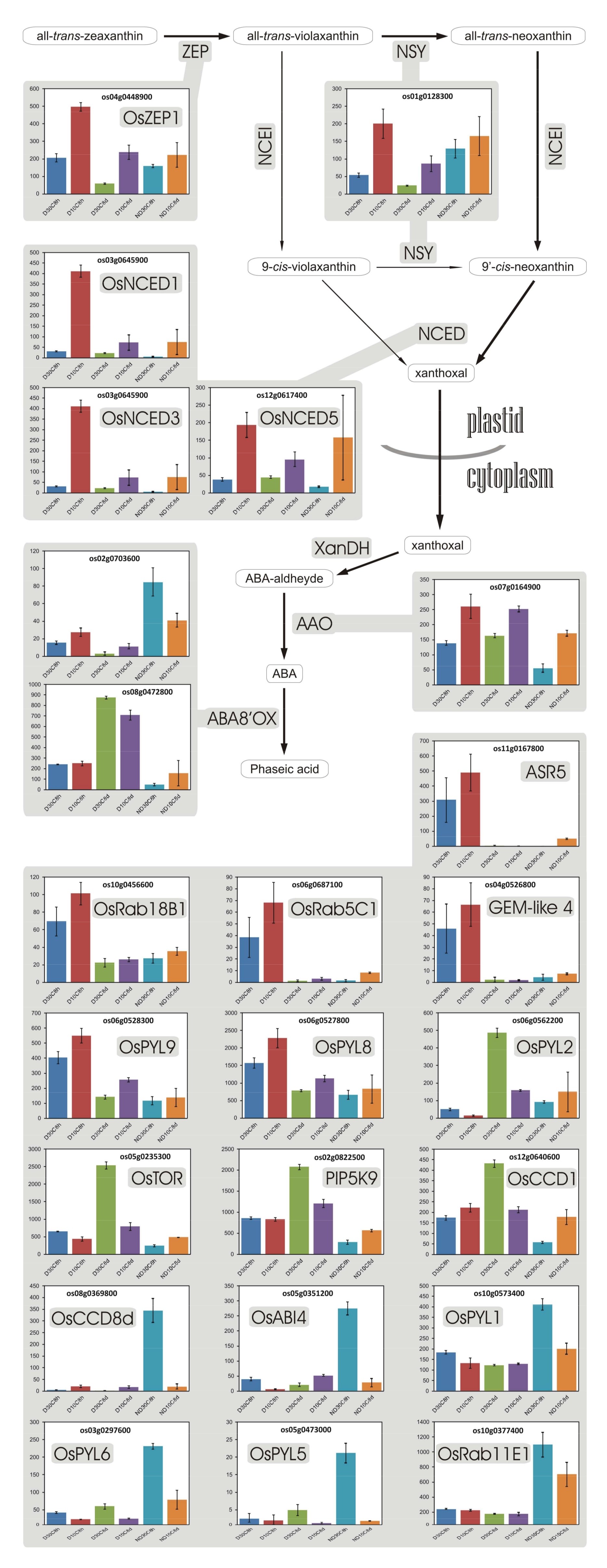
3.11. Abscisic Acid
3.12. Gibberellins
3.13. Seed Storage Proteins
3.14. Soluble Starch Synthase
3.15. Pre-Emptive Defence Strategies and Regulation of Transcription
3.16. More on Transcription Factors
3.17. Gene Co-Expression Network Analysis and the Role of Photosynthesis-Related Transcripts
3.18. Long Non-Coding RNAs
4. Materials and Methods
4.1. Seed Materials and Experimental Setup
4.2. RNA Extraction, Libraries Preparation and Sequencing
4.3. Bioinformatic and Statistical Methods
4.4. DEG Calling
4.5. Screening DEGs for Biological Functions
4.6. GO Term Enrichment Analyses
4.7. Co-Expression Analyses
4.8. Quantitative RT-PCR Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| D | dormant |
| ND | non-dormant |
| DEG | differentially expressed gene |
| FC | fold change |
| GO | Gene Ontology |
| PAs | proanthocyanidins |
| JA | jasmonate |
| GA | gibberellin |
| ABA | abscisic acid |
References
- Ziska, L.H.; Gealy, D.R.; Burgos, N.; Caicedo, A.L.; Gressel, J.; Lawton-Rauh, A.L.; Avila, L.A.; Theisen, G.; Norsworthy, J.; Ferrero, A.; et al. Weedy (red) rice: An emerging constraint to global rice production. Adv. Agron. 2015, 129, 181–228. [Google Scholar] [CrossRef]
- Bewley, J.D.; Bradford, K.J.; Hilhorst, H.W.M.; Nonogaki, H. Seeds: Physiology of Development, Germination and Dormancy, 3rd ed.; Springer: New York, NY, USA, 2013; ISBN 978-1-4614-4692-7. [Google Scholar]
- Finch-Savage, W.E.; Leubner-Metzger, G. Seed dormancy and the control of germination. New Phytol. 2006, 171, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth, M.J.; Bentsink, L.; Soppe, W.J.J. Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol. 2008, 179, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Footitt, S.; Cohn, M.A. Seed dormancy in red rice (Oryza sativa). IX. Embryo fructose-2,6-bisphosphate during dormancy breaking and subsequent germination. Plant Physiol. 1995, 107, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Gianinetti, A. Anomalous germination of dormant dehulled red rice seeds provides a new perspective to study the transition from dormancy to germination and to unravel the role of the caryopsis coat in seed dormancy. Seed Sci. Res. 2016, 26, 124–138. [Google Scholar] [CrossRef]
- Leopold, A.C.; Glenister, R.; Cohn, M.A. Relationship between water content and afterripening in red rice. Physiol. Plant. 1988, 74, 659–662. [Google Scholar] [CrossRef]
- Gianinetti, A.; Cohn, M.A. Seed dormancy in red rice. XII. Population-based analysis of dry-afterripening with a hydrotime model. Seed Sci. Res. 2007, 17, 253–271. [Google Scholar] [CrossRef]
- Gianinetti, A.; Cohn, M.A. Seed dormancy in red rice. XIII. Interaction of dry-afterripening and hydration temperature. Seed Sci. Res. 2008, 18, 151–159. [Google Scholar] [CrossRef]
- Carrera, E.; Holman, T.; Medhurst, A.; Dietrich, D.; Footitt, S.; Theodoulou, F.L.; Holdsworth, M.J. Seed after-ripening is a discrete developmental pathway associated with specific gene networks in Arabidopsis. Plant J. 2008, 53, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N. The relation of water absorption to germination of rice seed. Sci. Rep. Res. Inst. Tohoku Univ. Ser. Agric. 1961, 12, 61–71. [Google Scholar]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Footitt, S.; Cohn, M.A. Seed dormancy in red rice. VIII. Embryo acidification during dormancy-breaking and subsequent germination. Plant Physiol. 1992, 100, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- An, Y.-Q.; Lin, L. Transcriptional regulatory programs underlying barley germination and regulatory functions of Gibberellin and abscisic acid. BMC Plant Biol. 2011, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Rajjou, L.; Duval, M.; Gallardo, K.; Catusse, J.; Bally, J.; Job, C.; Job, D. Seed germination and vigor. Annu. Rev. Plant Biol. 2012, 63, 507–533. [Google Scholar] [CrossRef] [PubMed]
- Galland, M.; Huguet, R.; Arc, E.; Cueff, G.; Job, D.; Rajjou, L. Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during Arabidopsis seed germination. Mol. Cell. Proteom. 2014, 13, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Née, G.; Xiang, Y.; Soppe, W.J. The release of dormancy, a wake-up call for seeds to germinate. Curr. Opin. Plant Biol. 2017, 35, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Weitbrecht, K.; Müller, K.; Leubner-Metzger, G. First off the mark: Early seed germination. J. Exp. Bot. 2011, 62, 3289–3309. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.T.; Ribone, P.A.; Chan, R.L.; Ligterink, W.; Hilhorst, H.W.M. A predictive coexpression network identifies novel genes controlling the seed-to-seedling phase transition in Arabidopsis thaliana. Plant Physiol. 2016, 170, 2218–2231. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.A.; Narsai, R.; Carroll, A.; Ivanova, A.; Lohse, M.; Usadel, B.; Millar, A.H.; Whelan, J. Mapping metabolic and transcript temporal switches during germination in rice highlights specific transcription factors and the role of RNA instability in the germination process. Plant Physiol. 2009, 149, 961–980. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Han, C.; Yao, J.; Shen, S.; Yang, P. Constructing the metabolic and regulatory pathways in germinating rice seeds through proteomic approach. Proteomics 2011, 11, 2693–2713. [Google Scholar] [CrossRef] [PubMed]
- Sano, N.; Permana, H.; Kumada, R.; Shinozaki, Y.; Tanabata, T.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Proteomic analysis of embryonic proteins synthesized from long-lived mRNAs during germination of rice seeds. Plant Cell Physiol. 2012, 53, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Novák, O.; Ljung, K.; Hanson, J.; Bentsink, L. Combined transcriptome and translatome analyses reveal a role for tryptophan-dependent auxin biosynthesis in the control of DOG1 -dependent seed dormancy. New Phytol. 2018, 217, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Gao, F.; Kanno, Y.; Jordan, M.C.; Kamiya, Y.; Seo, M.; Ayele, B.T. Regulation of wheat seed dormancy by after-ripening is mediated by specific transcriptional switches that induce changes in seed hormone metabolism and signaling. PLoS ONE 2013, 8, e56570. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, K.L.; Ottis, B.V.; Prazak-Havey, A.M.; Bormans, C.A.; Sneller, C.; Chandler, J.M.; Park, W.D. Is all red rice found in commercial rice really Oryza sativa? Weed Sci. 2001, 49, 468–476. [Google Scholar] [CrossRef]
- Usadel, B.; Nagel, A.; Steinhauser, D.; Gibon, Y.; Bläsing, O.E.; Redestig, H.; Sreenivasulu, N.; Krall, L.; Hannah, M.A.; Poree, F.; et al. PageMan: An interactive ontology tool to generate, display, and annotate overview graphs for profiling experiments. BMC Bioinform. 2006, 7, 535. [Google Scholar] [CrossRef] [PubMed]
- Usadel, B.; Nagel, A.; Thimm, O.; Redestig, H.; Blaesing, O.E.; Palacios-Rojas, N.; Selbig, J.; Hannemann, J.; Piques, M.C.; Steinhauser, D.; et al. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 2005, 138, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Usadel, B.; Poree, F.; Nagel, A.; Lohse, M.; Czedik-Eysenberg, A.; Stitt, M. A guide to using MapMan to visualize and compare Omics data in plants: A case study in the crop species, Maize. Plant Cell Environ. 2009, 32, 1211–1229. [Google Scholar] [CrossRef] [PubMed]
- Arc, E.; Chibani, K.; Grappin, P.; Jullien, M.; Godin, B.; Cueff, G.; Valot, B.; Balliau, T.; Job, D.; Rajjou, L. Cold stratification and exogenous nitrates entail similar functional proteome adjustments during Arabidopsis seed dormancy release. J. Proteome Res. 2012, 11, 5418–5432. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.K.; Ariizumi, T.; Steber, C.M. Biology in the dry seed: Transcriptome changes associated with dry seed dormancy and dormancy loss in the Arabidopsis GA-insensitive sleepy1-2 mutant. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Botha, F.C.; Potgieter, G.P.; Botha, A.-M. Respiratory metabolism and gene expression during seed germination. Plant Growth Regul. 1992, 11, 211–224. [Google Scholar] [CrossRef]
- Menegus, F.; Cattaruzza, L.; Molinari, H.; Ragg, E. Rice and wheat seedlings as plant models of high and low tolerance to anoxia. In Surviving Hypoxia: Mechanisms of Control and Adaptation; Hochachka, P.W., Lutz, P.L., Sick, T., Rosenthal, M., van den Thillart, G., Eds.; CRC Press: Boca Raton, FL, USA, 1993; pp. 53–64. [Google Scholar]
- Yang, P.; Li, X.; Wang, X.; Chen, H.; Chen, F.; Shen, S. Proteomic analysis of rice (Oryza sativa) seeds during germination. Proteomics 2007, 7, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yamane, M.; Yamaji, N.; Kanamori, H.; Tagiri, A.; Schwerdt, J.G.; Fincher, G.B.; Matsumoto, T.; Takeda, K.; Komatsuda, T. Alanine aminotransferase controls seed dormancy in barley. Nat. Commun. 2016, 7, 11625. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.K.M.; Hooks, M.A.; Miaullis, A.P.; Edwards, S.; Webster, C. Contribution of malate and amino acid metabolism to cytoplasmic pH regulation in hypoxic maize root tips studied using nuclear magnetic resonance spectroscopy. Plant Physiol. 1992, 98, 480–487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Magneschi, L.; Perata, P. Rice germination and seedling growth in the absence of oxygen. Ann. Bot. 2009, 103, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Shingaki-Wells, R.N.; Huang, S.; Taylor, N.L.; Carroll, A.J.; Zhou, W.; Millar, A.H. Differential molecular responses of rice and wheat coleoptiles to anoxia reveal novel metabolic adaptations in amino acid metabolism for tissue tolerance. Plant Physiol. 2011, 156, 1706–1724. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yin, X.; He, D.; Yang, P. Analysis of proteome profile in germinating soybean seed, and its comparison with rice showing the styles of reserves mobilization in different crops. PLoS ONE 2013, 8, e56947. [Google Scholar] [CrossRef] [PubMed]
- Reggiani, R.; Cantu, C.A.; Brambilla, I.; Bertani, A. Accumulation and interconversion of amino acids in rice roots under anoxia. Plant Cell Physiol. 1988, 29, 981–987. [Google Scholar] [CrossRef]
- Jones, R.L.; Jacobsen, J.V. Regulation of synthesis and transport of secreted proteins in cereal aleurone. Int. Rev. Cytol. 1991, 126, 49–88. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.A.; Wilkinson, M.C.; Laidman, D.L. Lipid metabolism in germinating cereals. In Lipids in Cereal Technology; Barnes, P.J., Ed.; Academic Press: London, UK, 1983; pp. 57–92. ISBN 978-0-12-079020-3. [Google Scholar]
- Ma, Z.; Marsolais, F.; Bernards, M.A.; Sumarah, M.W.; Bykova, N.V.; Igamberdiev, A.U. Glyoxylate cycle and metabolism of organic acids in the scutellum of barley seeds during germination. Plant Sci. 2016, 248, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bewley, J.D. Seed germination and reserve mobilization. In eLS, Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2001; ISBN 978-0-470-01617-6. [Google Scholar]
- Penfield, S.; Rylott, E.L.; Gilday, A.D.; Graham, S.; Larson, T.R.; Graham, I.A. Reserve mobilization in the Arabidopsis endosperm fuels hypocotyl elongation in the dark, is independent of abscisic acid, and requires PHOSPHOENOLPYRUVATE CARBOXYKINASE1. Plant Cell 2004, 16, 2705–2718. [Google Scholar] [CrossRef] [PubMed]
- Graham, I.A. Seed storage oil mobilization. Annu. Rev. Plant Biol. 2008, 59, 115–142. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Beevers, H. Subcellular distribution of gluconeogenetic enzymes in germinating castor bean endosperm. Plant Physiol. 1979, 64, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Plaxton, W.C. The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 185–214. [Google Scholar] [CrossRef] [PubMed]
- Flores-Tornero, M.; Anoman, A.D.; Rosa-Téllez, S.; Toujani, W.; Weber, A.P.M.; Eisenhut, M.; Kurz, S.; Alseekh, S.; Fernie, A.R.; Muñoz-Bertomeu, J.; et al. Overexpression of the triose phosphate translocator (TPT) complements the abnormal metabolism and development of plastidial glycolytic glyceraldehyde-3-phosphate dehydrogenase mutants. Plant J. 2017, 89, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Rylott, E.L.; Gilday, A.D.; Graham, I.A. The gluconeogenic enzyme phosphoenolpyruvate carboxykinase in Arabidopsis is essential for seedling establishment. Plant Physiol. 2003, 131, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Eastmond, P.J.; Astley, H.M.; Parsley, K.; Aubry, S.; Williams, B.P.; Menard, G.N.; Craddock, C.P.; Nunes-Nesi, A.; Fernie, A.R.; Hibberd, J.M. Arabidopsis uses two gluconeogenic gateways for organic acids to fuel seedling establishment. Nat. Commun. 2015, 6, 6659. [Google Scholar] [CrossRef] [PubMed]
- Oaks, A.; Beevers, H. The glyoxylate cycle in maize scutellum. Plant Physiol. 1964, 39, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Holtman, W.L.; Heistek, J.C.; Mattern, K.A.; Bakhuizen, R.; Douma, A.C. β-oxidation of fatty acids is linked to the glyoxylate cycle in the aleurone but not in the embryo of germinating barley. Plant Sci. 1994, 99, 43–53. [Google Scholar] [CrossRef]
- Ratajczak, W.; Polcyn, W.; Lehmann, T.; Ratajczak, L.; Garnczarska, M. Metabolism of amino acids in germinating yellow lupin seeds. II. Pathway of conversion of aspartate to alanine during the imbibition. Acta Physiol. Plant. 1998, 20, 123–127. [Google Scholar] [CrossRef]
- Atwell, B.J.; Greenway, H.; Colmer, T.D. Efficient use of energy in anoxia-tolerant plants with focus on germinating rice seedlings. New Phytol. 2015, 206, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Ferjani, A.; Segami, S.; Horiguchi, G.; Muto, Y.; Maeshima, M.; Tsukaya, H. Keep an eye on PPi: The vacuolar-type H+-pyrophosphatase regulates postgerminative development in Arabidopsis. Plant Cell 2011, 23, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, G.; Lv, D.; Hu, Y.; Li, J.; Li, X.; Yan, Y. Transcriptome analysis during seed germination of elite Chinese bread wheat cultivar Jimai 20. BMC Plant Biol. 2014, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Itoh, H.; Ueguchi-Tanaka, M.; Ashikari, M.; Matsuoka, M. The α-amylase induction in endosperm during rice seed germination is caused by gibberellin synthesized in epithelium. Plant Physiol. 2002, 128, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Linares, L.; Gavilanes-Ruíz, M.; Díaz-Pontones, D.; Guzmán-Chávez, F.; Calzada-Alejo, V.; Zurita-Villegas, V.; Luna-Loaiza, V.; Moreno-Sánchez, R.; Bernal-Lugo, I.; Sánchez-Nieto, S. Early carbon mobilization and radicle protrusion in maize germination. J. Exp. Bot. 2012, 63, 4513–4526. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Xu, X.; Zhang, L.; Zhang, H.; Lin, L.; Wang, Q.; Li, Q.; Ge, S.; Lu, B.-R.; Wang, W.; et al. Duplication and independent selection of cell-wall invertase genes GIF1 and OsCIN1 during rice evolution and domestication. BMC Evol. Biol. 2010, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Ricard, B.; Rivoal, J.; Spiteri, A.; Pradet, A. Anaerobic stress induces the transcription and translation of sucrose synthase in rice. Plant Physiol. 1991, 95, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.T.; Licausi, F. Oxygen sensing and signaling. Annu. Rev. Plant Biol. 2015, 66, 345–367. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-J.; Xu, H.-H.; Wang, W.-Q.; Li, N.; Wang, W.-P.; Lu, Z.; Møller, I.M.; Song, S.-Q. Identification of embryo proteins associated with seed germination and seedling establishment in germinating rice seeds. J. Plant Physiol. 2016, 196–197, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Scofield, G.N.; Terao, T. An expression analysis profile for the entire sucrose synthase gene family in rice. Plant Sci. 2008, 174, 534–543. [Google Scholar] [CrossRef]
- Huang, S.; Greenway, H.; Colmer, T.D.; Millar, A.H. Protein synthesis by rice coleoptiles during prolonged anoxia: Implications for glycolysis, growth and energy utilization. Ann. Bot. 2005, 96, 703–715. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lasanthi-Kudahettige, R.; Magneschi, L.; Loreti, E.; Gonzali, S.; Licausi, F.; Novi, G.; Beretta, O.; Vitulli, F.; Alpi, A.; Perata, P. Transcript profiling of the anoxic rice coleoptile. Plant Physiol. 2007, 144, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Larondelle, Y.; Corbineau, F.; Dethier, M.; Come, D.; Hers, H.-G. Fructose 2,6-bisphosphate in germinating oat seeds. A biochemical study of seed dormancy. Eur. J. Biochem. 1987, 166, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Heineke, D.; Riens, B.; Grosse, H.; Hoferichter, P.; Peter, U.; Flugge, U.-I.; Heldt, H.W. Redox transfer across the inner chloroplast envelope membrane. Plant Physiol. 1991, 95, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Sechet, J.; Frey, A.; Cuzzi, D.; Berger, A.; Perreau, F.; Cueff, G.; Charif, D.; Rajjou, L.; Mouille, G.; North, H.M.; et al. Xyloglucan metabolism differentially impacts the cell wall characteristics of the endosperm and embryo during Arabidopsis seed germination. Plant Physiol. 2016, 170, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Voegele, A.; Linkies, A.; Müller, K.; Leubner-Metzger, G. Members of the gibberellin receptor gene family GID1 (GIBBERELLIN INSENSITIVE DWARF1) play distinct roles during Lepidium sativum and Arabidopsis thaliana seed germination. J. Exp. Bot. 2011, 62, 5131–5147. [Google Scholar] [CrossRef] [PubMed]
- Nonogaki, H.; Chen, F.; Bradford, K.J. Mechanisms and genes involved in germination sensu stricto. In Seed Development, Dormancy and Germination; Bradford, K.J., Nonogaki, H., Eds.; Blackwell Publishing Ltd.: Oxford, UK, 2007; pp. 264–304. ISBN 978-0-470-98884-8. [Google Scholar]
- Wei, T.; He, Z.; Tan, X.; Liu, X.; Yuan, X.; Luo, Y.; Hu, S. An integrated RNA-Seq and network study reveals a complex regulation process of rice embryo during seed germination. Biochem. Biophys. Res. Commun. 2015, 464, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Usadel, B.; Winter, A.; Radchuk, V.; Scholz, U.; Stein, N.; Weschke, W.; Strickert, M.; Close, T.J.; Stitt, M.; et al. Barley grain maturation and germination: Metabolic pathway and regulatory network commonalities and differences highlighted by new MapMan/PageMan profiling tools. Plant Physiol. 2008, 146, 1738–1758. [Google Scholar] [CrossRef] [PubMed]
- Barrero, J.M.; Talbot, M.J.; White, R.G.; Jacobsen, J.V.; Gubler, F. Anatomical and transcriptomic studies of the coleorhiza reveal the importance of this tissue in regulating dormancy in barley. Plant Physiol. 2009, 150, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Geilfus, C.-M.; Zörb, C.; Neuhaus, C.; Hansen, T.; Lüthen, H.; Mühling, K.H. Differential transcript expression of wall-loosening candidates in leaves of maize cultivars differing in salt resistance. J. Plant Growth Regul. 2011, 30, 387–395. [Google Scholar] [CrossRef]
- Chen, F.; Nonogaki, H.; Bradford, K.J. A gibberellin-regulated xyloglucan endotransglycosylase gene is expressed in the endosperm cap during tomato seed germination. J. Exp. Bot. 2002, 53, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.; Linkies, A.; Muller, K.; Oracz, K.; Wang, X.; Lynn, J.R.; Leubner-Metzger, G.; Finch-Savage, W.E. Regulation of seed germination in the close Arabidopsis relative Lepidium sativum: A global tissue-specific transcript analysis. Plant Physiol. 2011, 155, 1851–1870. [Google Scholar] [CrossRef] [PubMed]
- Bellieny-Rabelo, D.; de Oliveira, E.A.G.; da Silva Ribeiro, E.; Costa, E.P.; Oliveira, A.E.A.; Venancio, T.M. Transcriptome analysis uncovers key regulatory and metabolic aspects of soybean embryonic axes during germination. Sci. Rep. 2016, 6, 36009. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Kermode, A.R. An increase in pectin methyl esterase activity accompanies dormancy breakage and germination of yellow cedar seeds. Plant Physiol. 2000, 124, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Scheler, C.; Weitbrecht, K.; Pearce, S.P.; Hampstead, A.; Büttner-Mainik, A.; Lee, K.J.D.; Voegele, A.; Oracz, K.; Dekkers, B.J.W.; Wang, X.; et al. Promotion of testa rupture during garden cress germination involves seed compartment-specific expression and activity of pectin methylesterases. Plant Physiol. 2015, 167, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Sitrit, Y.; Hadfield, K.A.; Bennett, A.B.; Bradford, K.J.; Downie, A.B. Expression of a polygalacturonase associated with tomato seed germination. Plant Physiol. 1999, 121, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Gianinetti, A.; Vernieri, P. On the role of abscisic acid in seed dormancy of red rice. J. Exp. Bot. 2007, 58, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, F.; Ferrari, B.; Gianinetti, A.; Dall’Asta, C.; Galaverna, G.; Scazzina, F.; Pellegrini, N. Characterization of antioxidant compounds of red and white rice and changes in total antioxidant capacity during processing. Mol. Nutr. Food Res. 2007, 51, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.-Y.; Foley, M.E.; Horvath, D.P.; Anderson, J.V.; Feng, J.; Zhang, L.; Mowry, C.R.; Ye, H.; Suttle, J.C.; Kadowaki, K.-I.; et al. Association between seed dormancy and pericarp color is controlled by a pleiotropic gene that regulates abscisic acid and flavonoid synthesis in weedy red rice. Genetics 2011, 189, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.H.; Chu, H.; Tang, L.K.; Sakamoto, W.; Maekawa, M.; Chu, I.K.; Wang, M.; Lo, C. Functional characterization of key structural genes in rice flavonoid biosynthesis. Planta 2008, 228, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Carrera, E.; Holman, T.; Medhurst, A.; Peer, W.; Schmuths, H.; Footitt, S.; Theodoulou, F.L.; Holdsworth, M.J. Gene expression profiling reveals defined functions of the ATP-binding cassette transporter COMATOSE late in phase II of germination. Plant Physiol. 2007, 143, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pang, Y.; Dixon, R.A. The mysteries of proanthocyanidin transport and polymerization. Plant Physiol. 2010, 153, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Dixon, R.A. The ‘ins’ and ‘outs’ of flavonoid transport. Trends Plant Sci. 2010, 15, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Memelink, J. Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 2016, 34, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-M.; Park, J.-Y.; Han, S.-H.; Anderson, A.J.; Yang, K.-Y.; Gardener, B.M.; Kim, Y.-C. Identification and transcriptional analysis of priming genes in Arabidopsis thaliana induced by root colonization with Pseudomonas chlororaphis O6. Plant Pathol. J. 2011, 27, 272–279. [Google Scholar] [CrossRef]
- Dey, S.; Vlot, A.C. Ethylene responsive factors in the orchestration of stress responses in monocotyledonous plants. Front. Plant Sci. 2015, 6, 640. [Google Scholar] [CrossRef] [PubMed]
- Gianinetti, A.; Laarhoven, L.J.J.; Persijn, S.T.; Harren, F.J.M.; Petruzzelli, L. Ethylene production is associated with germination but not seed dormancy in red rice. Ann. Bot. 2007, 99, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in Annals of Botany. Ann. Bot. 2013, 111, 1021–1058. [Google Scholar] [CrossRef] [PubMed]
- Dave, A.; Hernández, M.L.; He, Z.; Andriotis, V.M.E.; Vaistij, F.E.; Larson, T.R.; Graham, I.A. 12-Oxo-phytodienoic acid accumulation during seed development represses seed germination in Arabidopsis. Plant Cell 2011, 23, 583–599. [Google Scholar] [CrossRef] [PubMed]
- Dave, A.; Vaistij, F.E.; Gilday, A.D.; Penfield, S.D.; Graham, I.A. Regulation of Arabidopsis thaliana seed dormancy and germination by 12-oxo-phytodienoic acid. J. Exp. Bot. 2016, 67, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Linkies, A.; Leubner-Metzger, G. Beyond gibberellins and abscisic acid: How ethylene and jasmonates control seed germination. Plant Cell Rep. 2012, 31, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Won, C.; Shen, X.; Mashiguchi, K.; Zheng, Z.; Dai, X.; Cheng, Y.; Kasahara, H.; Kamiya, Y.; Chory, J.; Zhao, Y. Conversion of tryptophan to indole-3-acetic acid by TRYPTOPHAN AMINOTRANSFERASES OF ARABIDOPSIS and YUCCAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 18518–18523. [Google Scholar] [CrossRef] [PubMed]
- Weijers, D.; Wagner, D. Transcriptional responses to the auxin hormone. Annu. Rev. Plant Biol. 2016, 67, 539–574. [Google Scholar] [CrossRef] [PubMed]
- Strader, L.C.; Zhao, Y. Auxin perception and downstream events. Curr. Opin. Plant Biol. 2016, 33, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-T.; Wu, K. Role of histone deacetylases HDA6 and HDA19 in ABA and abiotic stress response. Plant Signal. Behav. 2010, 5, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Abu-Zaitoon, Y.M.; Bennett, K.; Normanly, J.; Nonhebel, H.M. A large increase in IAA during development of rice grains correlates with the expression of tryptophan aminotransferase OsTAR1 and a grain-specific YUCCA. Physiol. Plant. 2012, 146, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Coneva, V.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Nambara, E.; Bi, Y.-M.; Rothstein, S.J. OsPIN5b modulates rice (Oryza sativa) plant architecture and yield by changing auxin homeostasis, transport and distribution. Plant J. 2015, 83, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Goggin, D.E.; Steadman, K.J.; Emery, R.J.N.; Farrow, S.C.; Benech-Arnold, R.L.; Powles, S.B. ABA inhibits germination but not dormancy release in mature imbibed seeds of Lolium rigidum Gaud. J. Exp. Bot. 2009, 60, 3387–3396. [Google Scholar] [CrossRef] [PubMed]
- Chibani, K.; Ali-Rachedi, S.; Job, C.; Job, D.; Jullien, M.; Grappin, P. Proteomic analysis of seed dormancy in Arabidopsis. Plant Physiol. 2006, 142, 1493–1510. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.; Liu, X.; Song, J.; Li, H.; Sui, Z.; Zhang, M.; Fang, S.; Chu, J.; Xin, M.; et al. Up-regulating the abscisic acid inactivation gene ZmABA8ox1b contributes to seed germination heterosis by promoting cell expansion. J. Exp. Bot. 2016, 67, 2889–2900. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sano, N.; Ono, H.; Murata, K.; Yamada, T.; Hirasawa, T.; Kanekatsu, M. Accumulation of long-lived mRNAs associated with germination in embryos during seed development of rice. J. Exp. Bot. 2015, 66, 4035–4046. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.A.; Jacobsen, J.V.; Ross, J.J.; Helliwell, C.A.; Poole, A.T.; Scofield, G.; Reid, J.B.; Gubler, F. Seed dormancy and ABA metabolism in Arabidopsis and barley: The role of ABA 8′-hydroxylase. Plant J. 2006, 45, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Chono, M. Field studies on the regulation of abscisic acid content and germinability during grain development of barley: Molecular and chemical analysis of pre-harvest sprouting. J. Exp. Bot. 2006, 57, 2421–2434. [Google Scholar] [CrossRef] [PubMed]
- Gubler, F.; Hughes, T.; Waterhouse, P.; Jacobsen, J. Regulation of dormancy in barley by blue light and after-ripening: Effects on abscisic acid and gibberellin metabolism. Plant Physiol. 2008, 147, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Hoang, H.H.; Sotta, B.; Gendreau, E.; Bailly, C.; Leymarie, J.; Corbineau, F. Water content: A key factor of the induction of secondary dormancy in barley grains as related to ABA metabolism. Physiol. Plant. 2013, 148, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Kushiro, T.; Okamoto, M.; Nakabayashi, K.; Yamagishi, K.; Kitamura, S.; Asami, T.; Hirai, N.; Koshiba, T.; Kamiya, Y.; Nambara, E. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: Key enzymes in ABA catabolism. EMBO J. 2004, 23, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Kuwahara, A.; Seo, M.; Kushiro, T.; Asami, T.; Hirai, N.; Kamiya, Y.; Koshiba, T.; Nambara, E. CYP707A1 and CYP707A2, which encode abscisic acid 8’-hydroxylases, are indispensable for proper control of seed dormancy and germination in Arabidopsis. Plant Physiol. 2006, 141, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.; Al-Babili, S. On the substrate specificity of the rice strigolactone biosynthesis enzyme DWARF27. Planta 2016, 243, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Nakashima, K.; Miyakawa, T.; Kuromori, T.; Tanokura, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular basis of the core regulatory network in ABA responses: Sensing, signaling and transport. Plant Cell Physiol. 2010, 51, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Abe, F.; Kawahigashi, H.; Nakazono, K.; Tagiri, A.; Matsumoto, T.; Utsugi, S.; Ogawa, T.; Handa, H.; Ishida, H.; et al. A wheat homolog of MOTHER OF FT AND TFL1 acts in the regulation of germination. Plant Cell 2011, 23, 3215–3229. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Cho, C.-W.; So, H.-A.; Kang, J.-S.; Chung, Y.S.; Lee, J.-H. Overexpression of VrUBC1, a mung bean E2 ubiquitin-conjugating enzyme, enhances osmotic stress tolerance in Arabidopsis. PLoS ONE 2013, 8, e66056. [Google Scholar] [CrossRef] [PubMed]
- Brioudes, F.; Thierry, A.-M.; Chambrier, P.; Mollereau, B.; Bendahmane, M. Translationally controlled tumor protein is a conserved mitotic growth integrator in animals and plants. Proc. Natl. Acad. Sci. USA 2010, 107, 16384–16389. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Moin, M.; Kumar, M.U.; Reddy, A.B.M.; Ren, M.; Datla, R.; Siddiq, E.A.; Kirti, P.B. Ectopic expression of Arabidopsis Target of Rapamycin (AtTOR) improves water-use efficiency and yield potential in rice. Sci. Rep. 2017, 7, 42835. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, A.; Citerne, S.; Jéhanno, I.; Bersimbaev, R.I.; Veit, B.; Meyer, C.; Leprince, A.-S. Mutations in the Arabidopsis Lst8 and Raptor genes encoding partners of the TOR complex, or inhibition of TOR activity decrease abscisic acid (ABA) synthesis. Biochem. Biophys. Res. Commun. 2015, 467, 992–997. [Google Scholar] [CrossRef] [PubMed]
- Hobo, T.; Kowyama, Y.; Hattori, T. A bZIP factor, TRAB1, interacts with VP1 and mediates abscisic acid-induced transcription. Proc. Natl. Acad. Sci. USA 1999, 96, 15348–15353. [Google Scholar] [CrossRef] [PubMed]
- Söderman, E.M.; Brocard, I.M.; Lynch, T.J.; Finkelstein, R.R. Regulation and function of the Arabidopsis ABA-insensitive4 gene in seed and abscisic acid response signaling networks. Plant Physiol. 2000, 124, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Penfield, S.; Li, Y.; Gilday, A.D.; Graham, S.; Graham, I.A. Arabidopsis ABA INSENSITIVE4 regulates lipid mobilization in the embryo and reveals repression of seed germination by the endosperm. Plant Cell 2006, 18, 1887–1899. [Google Scholar] [CrossRef] [PubMed]
- Wind, J.J.; Peviani, A.; Snel, B.; Hanson, J.; Smeekens, S.C. ABI4: Versatile activator and repressor. Trends Plant Sci. 2013, 18, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Richly, E.; Dietzmann, A.; Biehl, A.; Kurth, J.; Laloi, C.; Apel, K.; Salamini, F.; Leister, D. Covariations in the nuclear chloroplast transcriptome reveal a regulatory master-switch. EMBO Rep. 2003, 4, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Koussevitzky, S.; Nott, A.; Mockler, T.C.; Hong, F.; Sachetto-Martins, G.; Surpin, M.; Lim, J.; Mittler, R.; Chory, J. Multiple signals from damaged chloroplasts converge on a common pathway to regulate nuclear gene expression. Science 2007, 316, 715–719. [Google Scholar] [CrossRef]
- Zhang, J.; Hill, D.R.; Sylvester, A.W. Diversification of the RAB guanosine triphosphatase family in dicots and monocots. J. Integr. Plant Biol. 2007, 49, 1129–1141. [Google Scholar] [CrossRef]
- Jeong, H.-J.; Jung, K.-H. Rice tissue-specific promoters and condition-dependent promoters for effective translational application. J. Integr. Plant Biol. 2015, 57, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Moon, S.-J.; Min, M.K.; Choi, E.-H.; Kim, J.-A.; Koh, E.Y.; Yoon, I.; Byun, M.-O.; Yoo, S.-D.; Kim, B.-G. Functional characterization and reconstitution of ABA signaling components using transient gene expression in rice protoplasts. Front. Plant Sci. 2015, 6, 614. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Urano, K.; Yoshiwara, K.; Morishita, Y.; Sakurai, N.; Suzuki, H.; Kojima, M.; Sakakibara, H.; Shibata, D.; Saito, K.; et al. Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol. 2014, 164, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lai, Y.; Wu, X.; Wu, G.; Guo, C. Overexpression of OsEm1 encoding a group I LEA protein confers enhanced drought tolerance in rice. Biochem. Biophys. Res. Commun. 2016, 478, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Woodger, F.; Jacobsen, J.V.; Gubler, F. Gibberellin action in germinated cereal grains. In Plant Hormones; Davies, P.J., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 221–240. ISBN 978-1-4020-2684-3. [Google Scholar]
- Leon, R.G.; Bassham, D.C.; Owen, M.D.K. Thermal and hormonal regulation of the dormancy-germination transition in Amaranthus tuberculatus seeds. Weed Res. 2007, 47, 335–344. [Google Scholar] [CrossRef]
- Miransari, M.; Smith, D.L. Plant hormones and seed germination. Environ. Exp. Bot. 2014, 99, 110–121. [Google Scholar] [CrossRef]
- Jacobsen, J.V.; Pearce, D.W.; Poole, A.T.; Pharis, R.P.; Mander, L.N. Abscisic acid, phaseic acid and gibberellin contents associated with dormancy and germination in barley. Physiol. Plant. 2002, 115, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.-F.; Yang, S.-Y.; Chen, K.-T.; Hsing, Y.-I.; Zeevaart, J.A.D.; Chen, L.-J.; Yu, S.-M. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 2008, 20, 2603–2618. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Z.; Xu, Y.; Joo, S.-H.; Kim, S.-K.; Xue, Z.; Xu, Z.; Wang, Z.; Chong, K. OsGSR1 is involved in crosstalk between gibberellins and brassinosteroids in rice. Plant J. 2009, 57, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Xu, H.; Ye, S.; Wang, S.; Li, L.; Zhang, S.; Wang, X. Gibberellic Acid-Stimulated Arabidopsis6 serves as an integrator of gibberellin, abscisic acid, and glucose signaling during seed germination in Arabidopsis. Plant Physiol. 2015, 169, 2288–2303. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Hanada, A.; Yamauchi, Y.; Kuwahara, A.; Kamiya, Y.; Yamaguchi, S. Gibberellin biosynthesis and response during Arabidopsis seed germination. Plant Cell 2003, 15, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, K.; Okamoto, M.; Koshiba, T.; Kamiya, Y.; Nambara, E. Genome-wide profiling of stored mRNA in Arabidopsis thaliana seed germination: Epigenetic and genetic regulation of transcription in seed. Plant J. 2005, 41, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Preston, J.; Tatematsu, K.; Kanno, Y.; Hobo, T.; Kimura, M.; Jikumaru, Y.; Yano, R.; Kamiya, Y.; Nambara, E. Temporal expression patterns of hormone metabolism genes during imbibition of Arabidopsis thaliana seeds: A comparative study on dormant and non-dormant accessions. Plant Cell Physiol. 2009, 50, 1786–1800. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Wu, M.; Yan, L.; Hu, R.; Ali, I.; Gan, Y. AtEXP2 is involved in seed germination and abiotic stress response in Arabidopsis. PLoS ONE 2014, 9, e85208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Zhang, W.; Wu, K.; Zheng, F.; Tian, L.; Liu, X.; Duan, J. Expression and functional analysis of the plant-specific histone deacetylase HDT701 in rice. Front. Plant Sci. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Dure, L.; Waters, L. Long-lived messenger RNA: Evidence from cotton seed germination. Science 1965, 147, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Potokina, E.; Sreenivasulu, N.; Altschmied, L.; Michalek, W.; Graner, A. Differential gene expression during seed germination in barley (Hordeum vulgare L.). Funct. Integr. Genom. 2002, 2, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chua, N.-H. Arabidopsis Decapping 5 is required for mRNA decapping, P-body formation, and translational repression during postembryonic development. Plant Cell 2009, 21, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Law, S.R.; Carrie, C.; Xu, L.; Whelan, J. In-depth temporal transcriptome profiling reveals a crucial developmental switch with roles for RNA processing and organelle metabolism that are essential for germination in Arabidopsis. Plant Physiol. 2011, 157, 1342–1362. [Google Scholar] [CrossRef] [PubMed]
- Narsai, R.; Howell, K.A.; Millar, A.H.; O’Toole, N.; Small, I.; Whelan, J. Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana. Plant Cell 2007, 19, 3418–3436. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.; Xu, H.; Zhu, Y.; Liu, Q.-Q.; Cai, X.-L. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J. Exp. Bot. 2013, 64, 3453–3466. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; He, D.; Li, M.; Yang, P. In-depth proteomic analysis of rice embryo reveals its important roles in seed germination. Plant Cell Physiol. 2014, 55, 1826–1847. [Google Scholar] [CrossRef] [PubMed]
- Matsukura, C.; Saitoh, T.; Hirose, T.; Ohsugi, R.; Perata, P.; Yamaguchi, J. Sugar uptake and transport in rice embryo. Expression of companion cell-specific sucrose transporter (OsSUT1) induced by sugar and light. Plant Physiol. 2000, 124, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Tuncel, A.; Kawaguchi, J.; Ihara, Y.; Matsusaka, H.; Nishi, A.; Nakamura, T.; Kuhara, S.; Hirakawa, H.; Nakamura, Y.; Cakir, B.; et al. The rice endosperm ADP-glucose pyrophosphorylase large subunit is essential for optimal catalysis and allosteric regulation of the heterotetrameric enzyme. Plant Cell Physiol. 2014, 55, 1169–1183. [Google Scholar] [CrossRef] [PubMed]
- Nayar, S.; Sharma, R.; Tyagi, A.K.; Kapoor, S. Functional delineation of rice MADS29 reveals its role in embryo and endosperm development by affecting hormone homeostasis. J. Exp. Bot. 2013, 64, 4239–4253. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.A.; Xie, D.-Y.; Sharma, S.B. Proanthocyanidins - a final frontier in flavonoid research? New Phytol. 2005, 165, 9–28. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, L.; Routaboul, J.; Cheynier, V.; Lepiniec, L.; Debeaujon, I. Flavonoid oxidation in plants: From biochemical properties to physiological functions. Trends Plant Sci. 2007, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhu, N.; Wang, X.; Yi, Q.; Zhu, D.; Lai, Y.; Zhao, Y. Analysis of rice Snf2 family proteins and their potential roles in epigenetic regulation. Plant Physiol. Biochem. 2013, 70, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Sarnowska, E.; Gratkowska, D.M.; Sacharowski, S.P.; Cwiek, P.; Tohge, T.; Fernie, A.R.; Siedlecki, J.A.; Koncz, C.; Sarnowski, T.J. The role of SWI/SNF chromatin remodeling complexes in hormone crosstalk. Trends Plant Sci. 2016, 21, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-L.; Huang, L.-F.; Fang, J.-C.; Yeh, C.-H.; Hong, C.-Y.; Wu, S.-J.; Lu, C.-A. Divergence of the expression and subcellular localization of CCR4-associated factor 1 (CAF1) deadenylase proteins in Oryza sativa. Plant Mol. Biol. 2014, 85, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhao, X.; Zhang, L.; Tang, T.; Lu, C.; Chen, G.; Wang, X.; Bu, C.; Zhao, X. RNA-seq profiling the transcriptome of secondary seed dormancy in canola (Brassica napus L.). Chin. Sci. Bull. 2014, 59, 4341–4351. [Google Scholar] [CrossRef]
- Kapoor, M.; Arora, R.; Lama, T.; Nijhawan, A.; Khurana, J.P.; Tyagi, A.K.; Kapoor, S. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genom. 2008, 9, 451. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Iida, K.; Tanaka, M.; Yamaguchi, K.; Mizuhashi, K.; Kim, J.-M.; Takahashi, S.; Kobayashi, N.; Shigenobu, S.; Shinozaki, K.; et al. Novel stress-inducible antisense RNAs of protein-coding loci are synthesized by RNA-dependent RNA polymerase. Plant Physiol. 2017, 175, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Nonogaki, H. Seed dormancy and germination—Emerging mechanisms and new hypotheses. Front. Plant Sci. 2014, 5, 233. [Google Scholar] [CrossRef] [PubMed]
- Demarsy, E.; Buhr, F.; Lambert, E.; Lerbs-Mache, S. Characterization of the plastid-specific germination and seedling establishment transcriptional programme. J. Exp. Bot. 2012, 63, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Kühn, K.; Yin, G.; Duncan, O.; Law, S.R.; Kubiszewski-Jakubiak, S.; Kaur, P.; Meyer, E.; Wang, Y.; Small, C.C.; Giraud, E.; et al. Decreasing electron flux through the cytochrome and/or alternative respiratory pathways triggers common and distinct cellular responses dependent on growth conditions. Plant Physiol. 2015, 167, 228–250. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Huang, Y.; Zhu, N.; Zhao, Y. The rice WUSCHEL-related homeobox genes are involved in reproductive organ development, hormone signaling and abiotic stress response. Gene 2014, 549, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Cui, Y. A GPU-accelerated algorithm for biclustering analysis and detection of condition-dependent coexpression network modules. Sci. Rep. 2017, 7, 4162. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, D.B.; Pomeranz, Y. Ultrastructure of the mature ungerminated rice (Oryza sativa) caryopsis. The caryopsis coat and the aleurone cells. Am. J. Bot. 1977, 64, 966–973. [Google Scholar] [CrossRef]
- Bechtel, D.B.; Pomeranz, Y. Ultrastructure of the mature ungerminated rice (Oryza sativa) caryopsis. The germ. Am. J. Bot. 1978, 65, 75–85. [Google Scholar] [CrossRef]
- Weinl, S.; Held, K.; Schlcking, K.; Steinhorst, L.; Kuhlgert, S.; Hippler, M.; Kudla, J. A plastid protein crucial for Ca2+-regulated stomatal responses. New Phytol. 2008, 179, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Arenhart, R.A.; De Lima, J.C.; Pedron, M.; Carvalho, F.E.L.; Da Silveira, J.A.G.; Rosa, S.B.; Caverzan, A.; Andrade, C.M.B.; SchüNemann, M.; Margis, R.; et al. Involvement of ASR genes in aluminium tolerance mechanisms in rice. Plant Cell Environ. 2013, 36, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Howell, K.A.; Millar, A.H.; Whelan, J. Ordered assembly of mitochondria during rice germination begins with promitochondrial structures rich in components of the protein import apparatus. Plant Mol. Biol. 2006, 60, 201–223. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Dayanandan, P. Structural and histochemical studies on grain-filling in the caryopsis of rice (Oryza sativa L.). J. Biosci. 2003, 28, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Harrak, H.; Lagrange, T.; Bisanz-Seyer, C.; Lerbs-Mache, S.; Mache, R. The expression of nuclear genes encoding plastid ribosomal proteins precedes the expression of chloroplast genes during early phases of chloroplast development. Plant Physiol. 1995, 108, 685–692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murcha, M.W.; Wang, Y.; Narsai, R.; Whelan, J. The plant mitochondrial protein import apparatus—The differences make it interesting. Biochim. Biophys. Acta 2014, 1840, 1233–1245. [Google Scholar] [CrossRef] [PubMed]
- Bassel, G.W.; Fung, P.; Chow, T.-F.F.; Foong, J.A.; Provart, N.J.; Cutler, S.R. Elucidating the germination transcriptional program using small molecules. Plant Physiol. 2008, 147, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.T.; Thomson, M.J.; Pfeil, B.E.; McCouch, S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 2006, 18, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Maekawa, M.; Oki, T.; Suda, I.; Iida, S.; Shimada, H.; Takamure, I.; Kadowaki, K. The Rc and Rd genes are involved in proanthocyanidin synthesis in rice pericarp. Plant J. 2006, 49, 91–102. [Google Scholar] [CrossRef] [PubMed]
- López-Gómez, R.; Gómez-Lim, M.A. A method for extracting intact RNA from fruits rich in polysaccharides using ripe mango mesocarp. HortScience 1992, 27, 440–442. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MapMan: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.T.; Grimes, M.; Kutlu, B.; Bot, J.J.; Galas, D.J. RCytoscape: Tools for exploratory network analysis. BMC Bioinform. 2013, 14, 217–315. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.T.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Yang, J.; Hu, K.; An, B.; Deng, X.; Li, Y. Reliable selection and holistic stability evaluation of reference genes for rice under 22 different experimental conditions. Appl. Biochem. Biotechnol. 2016, 179, 753–775. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test | D | ND | ||
|---|---|---|---|---|
| ps | S1 | ps | S1 | |
| (%) | (%) | (%) | (%) | |
| 8 h 30 °C | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| 8 d 30 °C | 0 ± 0 | 0 ± 0 | 99 ± 1 | 99 ± 1 |
| 14 d 30 °C | 1 ± 1 | 1 ± 1 | 100 ± 0 | 100 ± 0 |
| 8 h 10 °C | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| 8 h 10 °C + 14 d 30 °C | 2 ± 1 | 2 ± 1 | 100 ± 0 | 100 ± 0 |
| 8 d 10 °C | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| 8 d 10 °C + 14 d 30 °C | 7 ± 5 | 7 ± 5 | 100 ± 0 | 100 ± 0 |
| Seed | Incubation Temperature (°C) | Time of Incubation | Total Number of Transcripts | Number of Non-Coding Transcripts |
|---|---|---|---|---|
| Dormant | 30 | 8 h | 32,355 | 3381 |
| 8 days | 28,865 | 2469 | ||
| 10 | 8 h | 30,378 | 2418 | |
| 8 days | 31,747 | 3214 | ||
| Non-Dormant | 30 | 8 h | 31,967 | 3116 |
| 10 | 8 days | 31,968 | 2899 |
| Comparison | DEGs | DEGs for Non-Coding Transcripts | Intent of the Comparison (Highlighted Differences) |
|---|---|---|---|
| D 30 °C 8 h vs. ND 30 °C 8 h | 3772 | 18 | Transition to germination during imbibition |
| D 10 °C 8 d vs. ND 10 °C 8 d | 92 | 0 | Transition to (potential) germination when metabolism has stabilized |
| D 30 °C 8 d vs. D 30 °C 8 h | 4468 | 24 | Stabilization of metabolism in D seeds at normal temperature |
| D 30 °C 8 d vs. D 10 °C 8 d | 5131 | 36 | Assessment of temperature effect in D seeds (stabilized metabolism) |
| D 30 °C 8 h vs. D 10 °C 8 h | 1299 | 4 | Assessment of temperature effect in D seeds (during imbibition) |
| D 10 °C 8 d vs. D 10 °C 8 h | 3192 | 9 | Stabilization of metabolism in D seeds at low temperature |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianinetti, A.; Finocchiaro, F.; Bagnaresi, P.; Zechini, A.; Faccioli, P.; Cattivelli, L.; Valè, G.; Biselli, C. Seed Dormancy Involves a Transcriptional Program That Supports Early Plastid Functionality during Imbibition. Plants 2018, 7, 35. https://doi.org/10.3390/plants7020035
Gianinetti A, Finocchiaro F, Bagnaresi P, Zechini A, Faccioli P, Cattivelli L, Valè G, Biselli C. Seed Dormancy Involves a Transcriptional Program That Supports Early Plastid Functionality during Imbibition. Plants. 2018; 7(2):35. https://doi.org/10.3390/plants7020035
Chicago/Turabian StyleGianinetti, Alberto, Franca Finocchiaro, Paolo Bagnaresi, Antonella Zechini, Primetta Faccioli, Luigi Cattivelli, Giampiero Valè, and Chiara Biselli. 2018. "Seed Dormancy Involves a Transcriptional Program That Supports Early Plastid Functionality during Imbibition" Plants 7, no. 2: 35. https://doi.org/10.3390/plants7020035
APA StyleGianinetti, A., Finocchiaro, F., Bagnaresi, P., Zechini, A., Faccioli, P., Cattivelli, L., Valè, G., & Biselli, C. (2018). Seed Dormancy Involves a Transcriptional Program That Supports Early Plastid Functionality during Imbibition. Plants, 7(2), 35. https://doi.org/10.3390/plants7020035