Imaging and Spectroscopy of Natural Fluorophores in Pine Needles


Abstract
1. Introduction
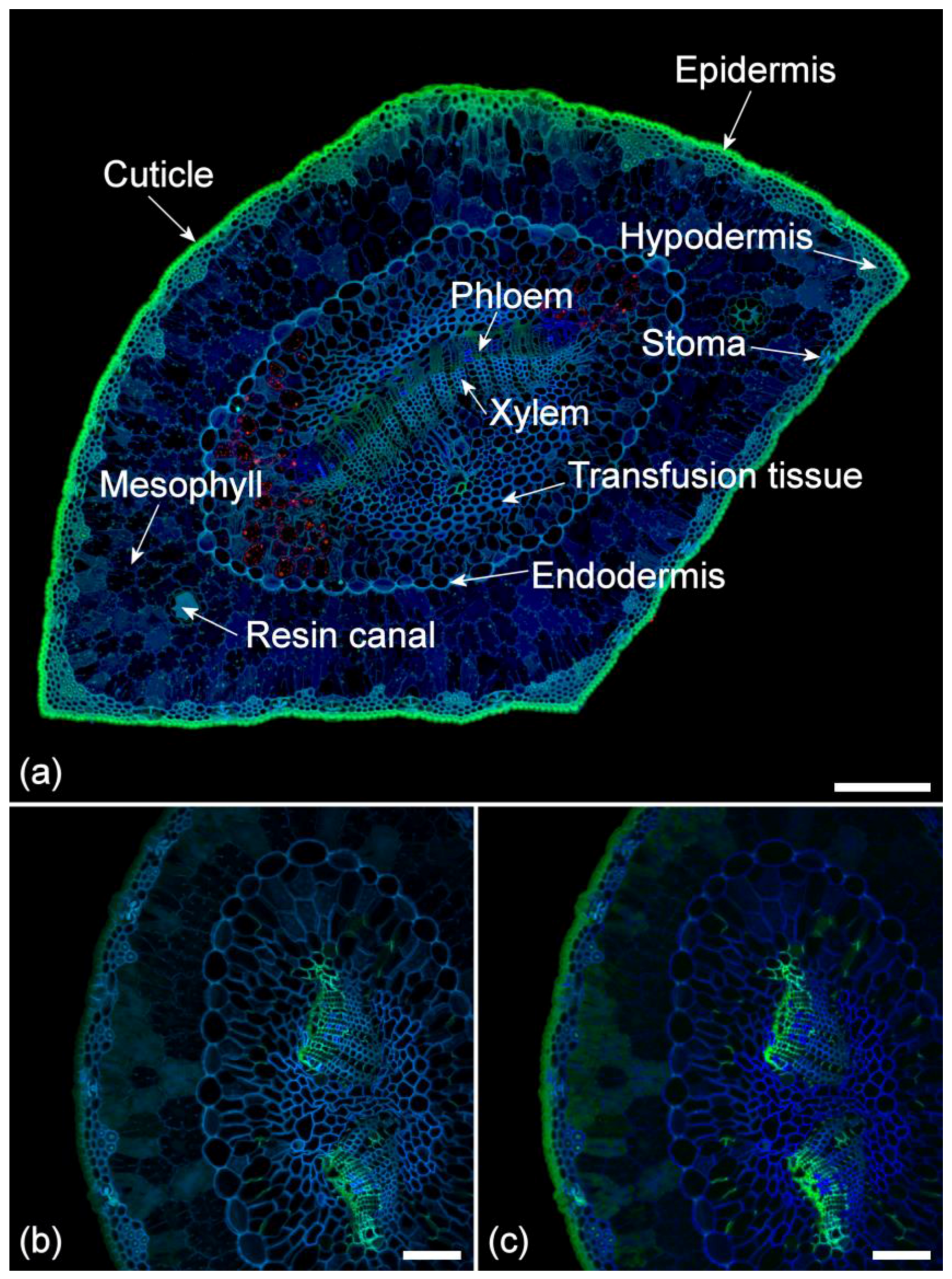
2. Results
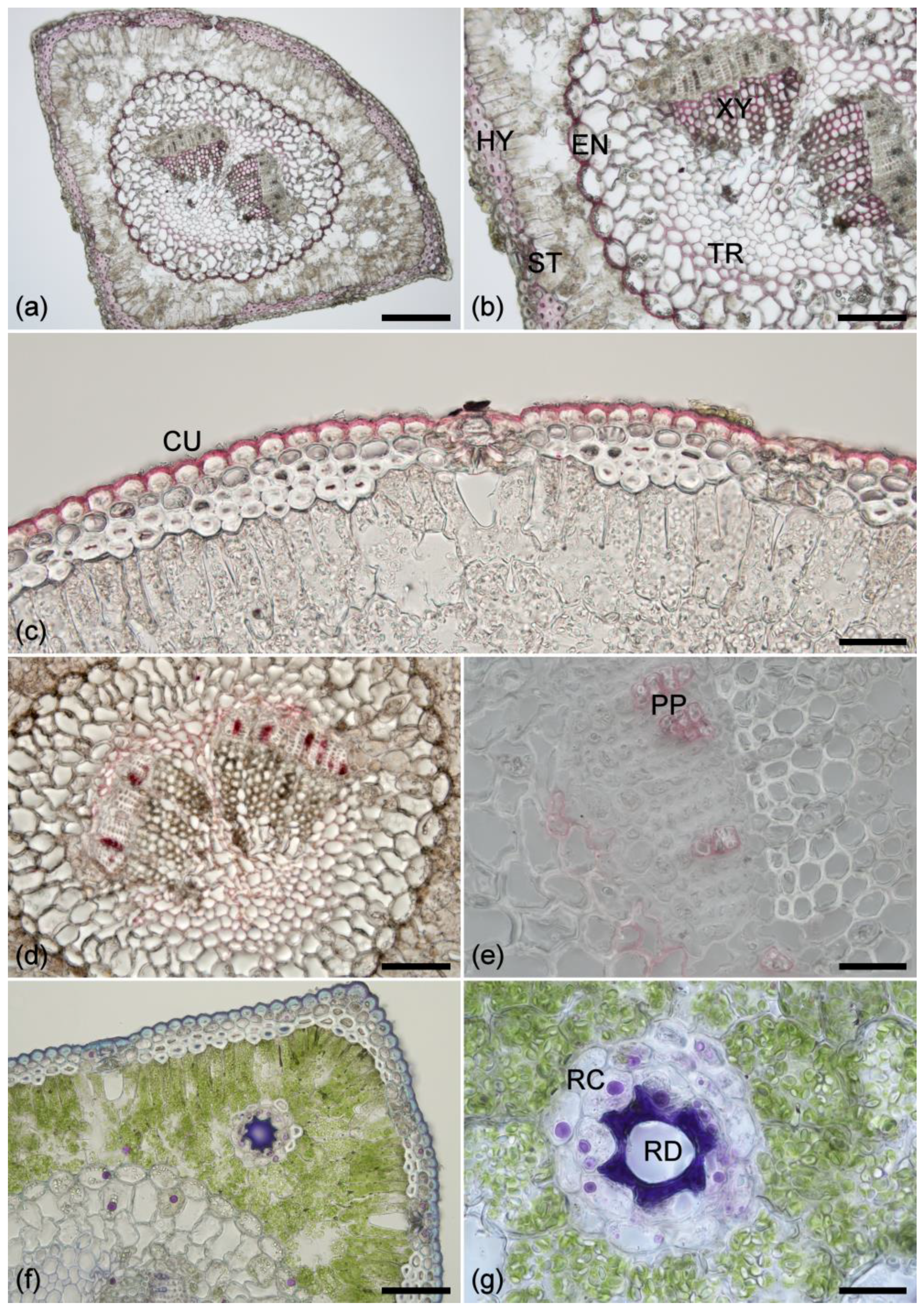
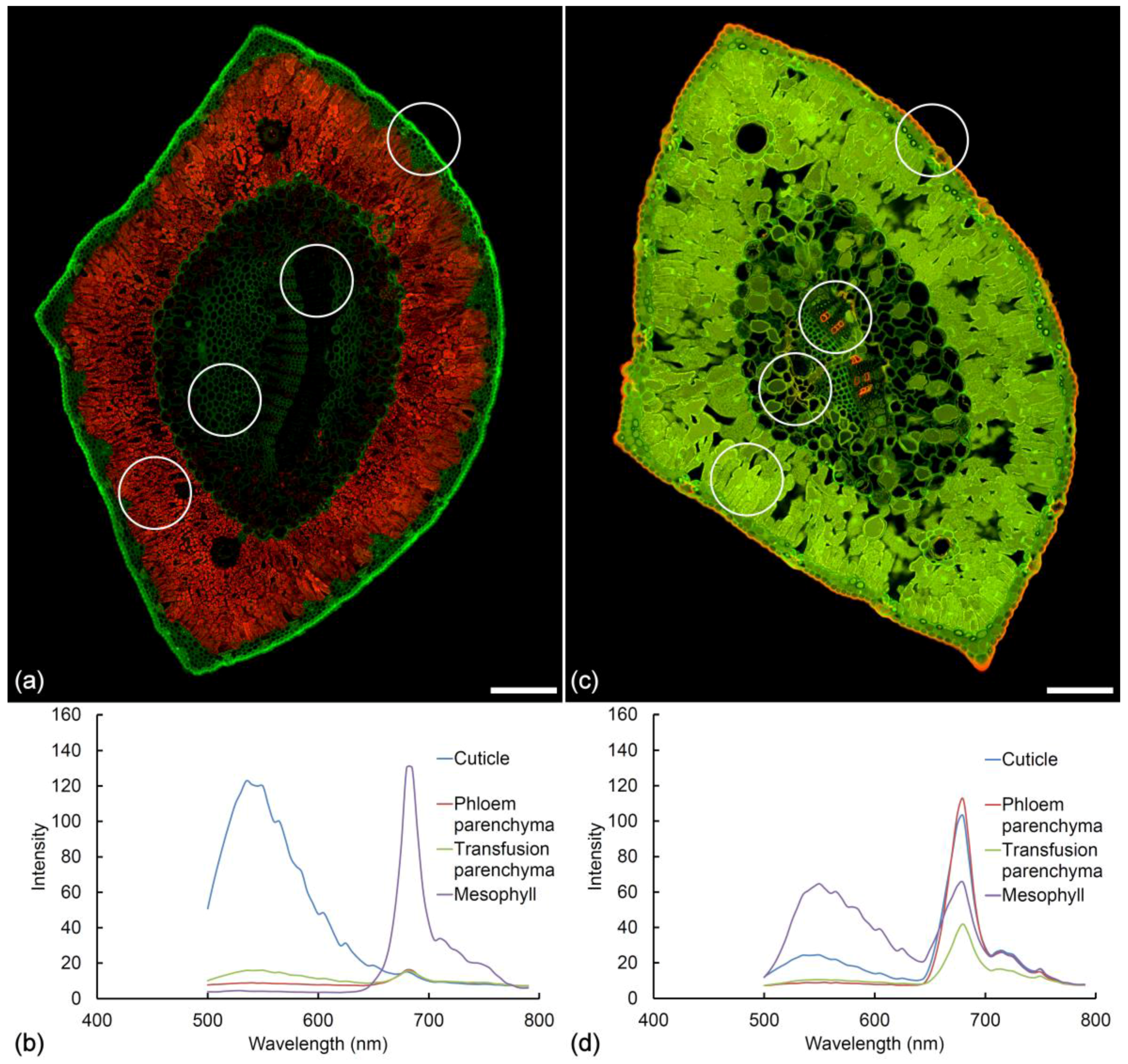
2.1. Histochemistry
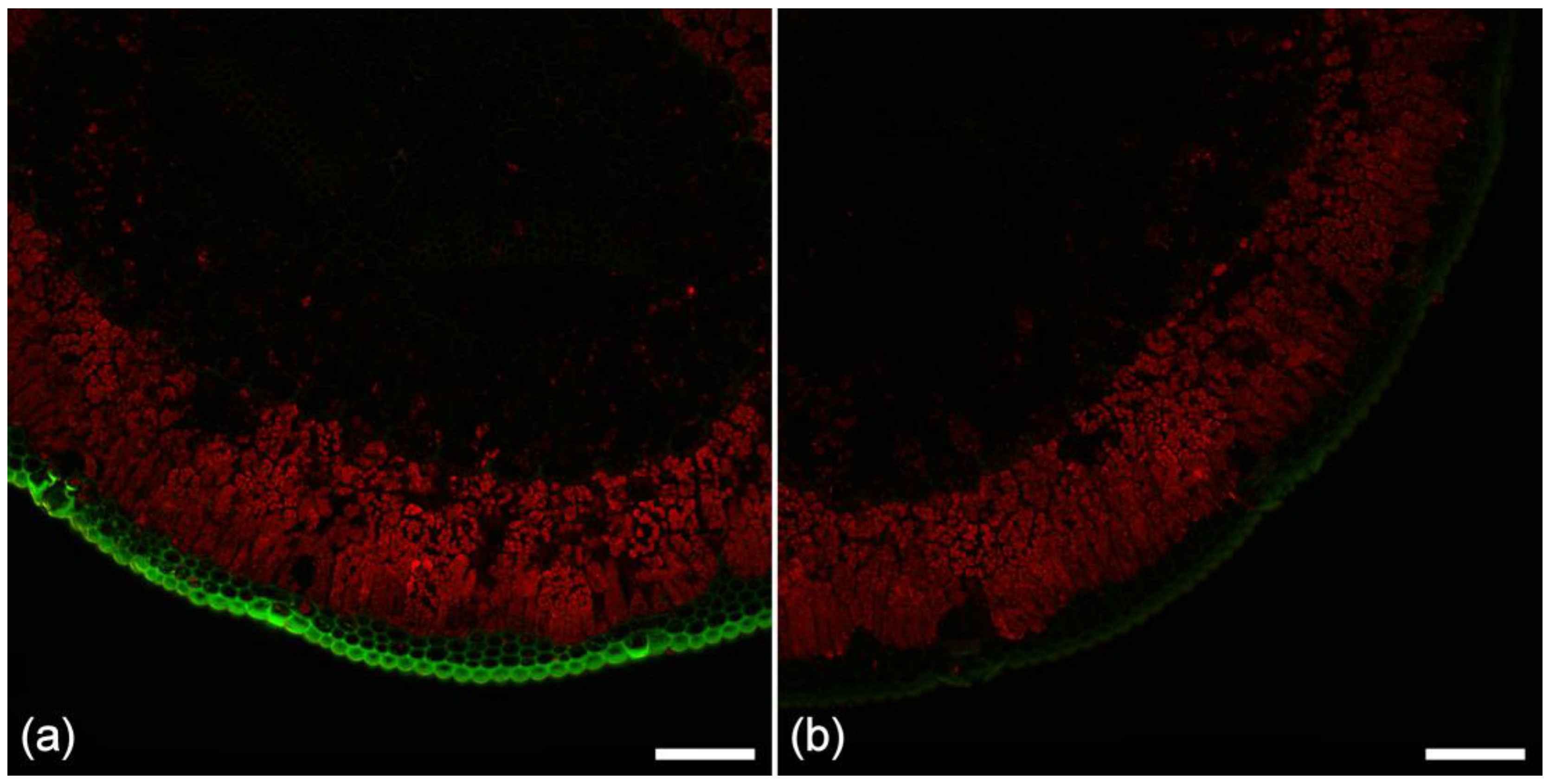
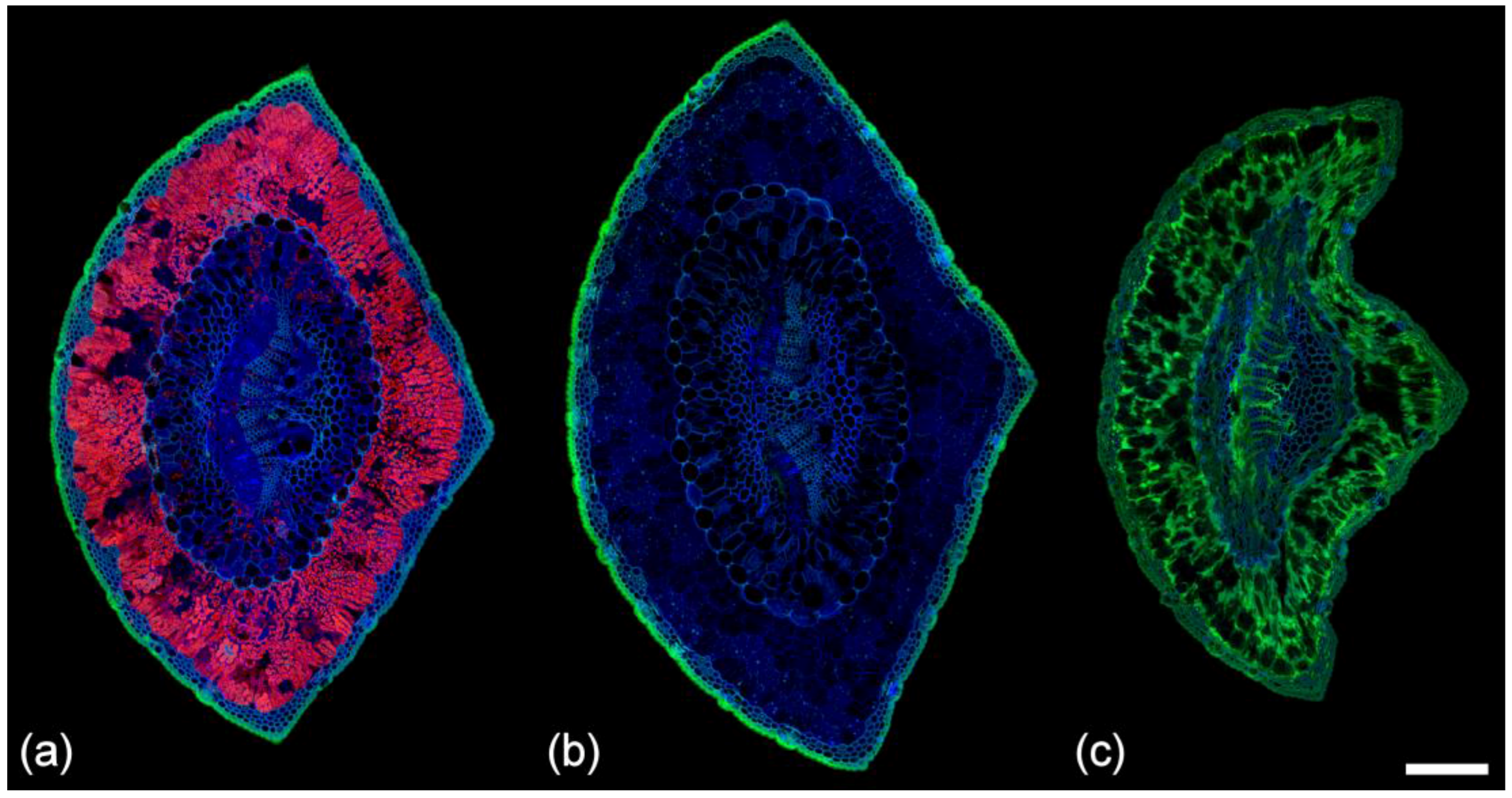
2.2. Lignin Fluorescence
2.3. Suberin Fluorescence
2.4. Ferulate Fluorescence
2.5. Cuticle Fluorescence
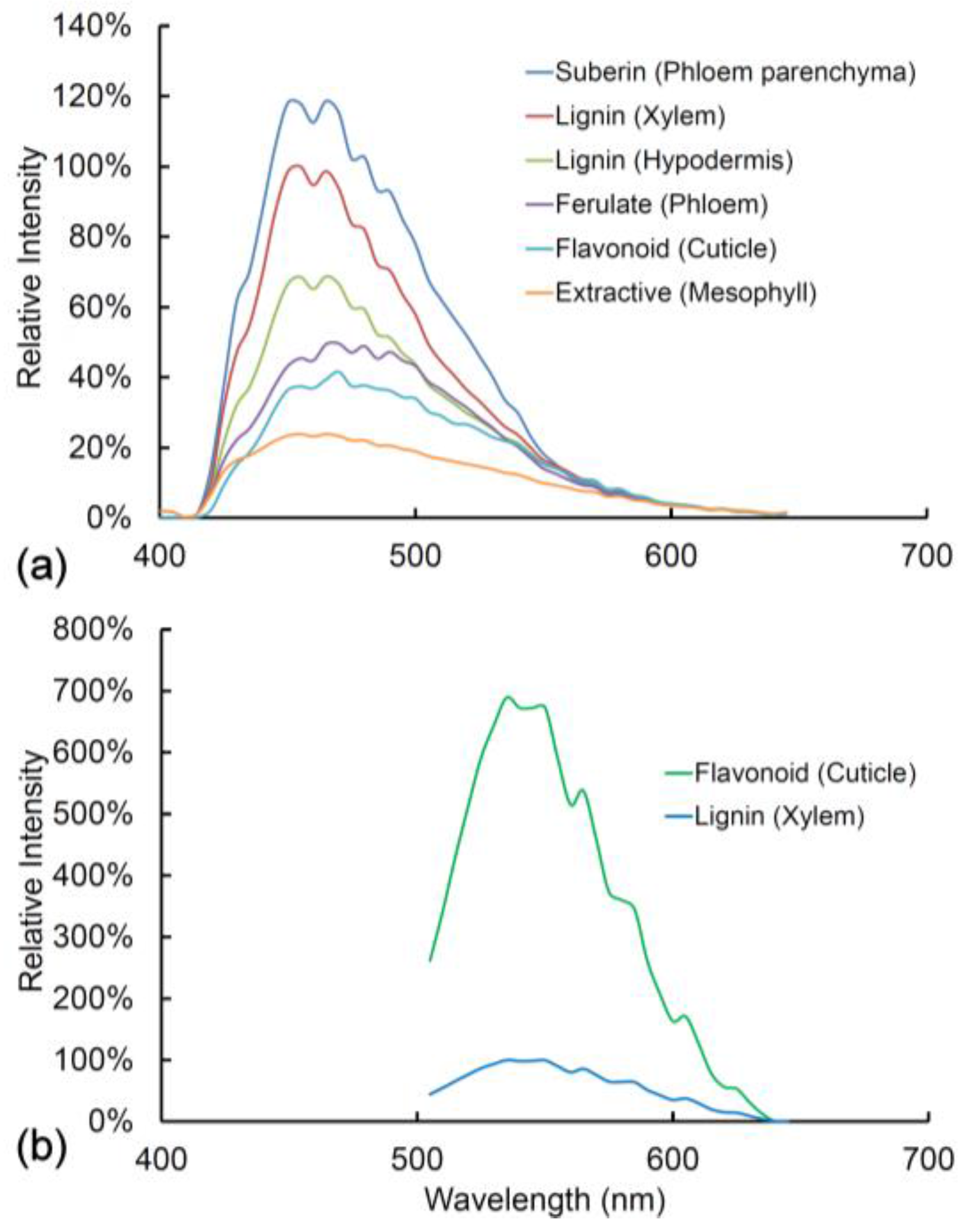
2.6. Other Cell Wall Fluorophores
2.7. Extractive Fluorescence
2.8. Chlorophyll Fluorescence
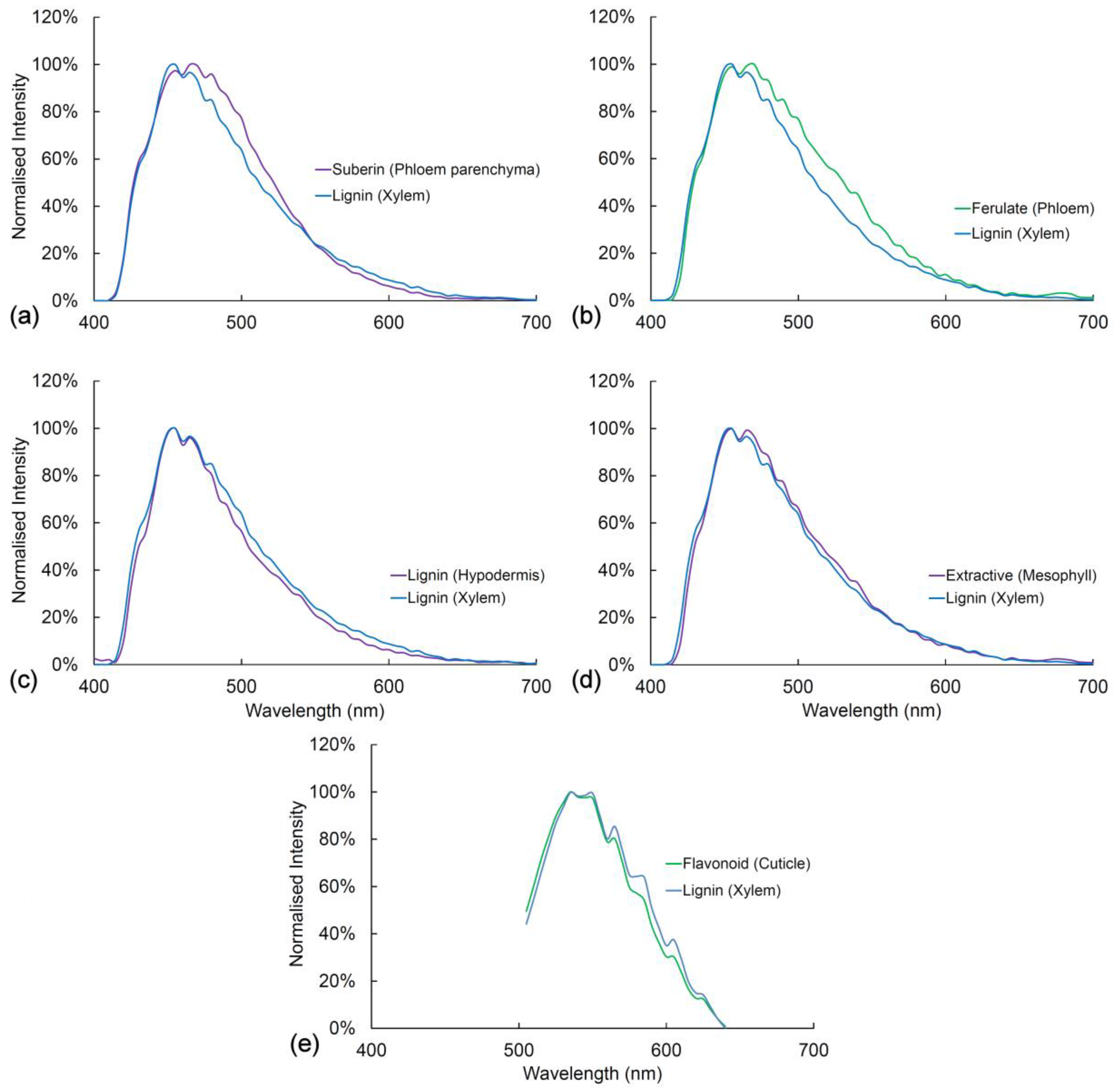
2.9. Comparison of Spectra
2.10. Fresh vs. Fixed Tissue
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Talamond, P.; Verdeil, J.-L.; Conéjéro, G. Secondary metabolite localization by autofluorescence in living plant cells. Molecules 2015, 20, 5024–5037. [Google Scholar] [CrossRef] [PubMed]
- Hutzler, P.; Fischbach, R.; Heller, W.; Jungblut, T.P.; Reuber, S.; Schmitz, R.; Veit, M.; Weissenböck, G.; Schnitzler, J.-P. Tissue localization of phenolic compounds in plants by confocal laser scanning microscopy. J. Exp. Bot. 1998, 49, 953–965. [Google Scholar] [CrossRef]
- Fernández, S.; Osorio, S.; Heredia, A. Monitoring and visualizing plant cuticles by confocal laser scanning microscopy. Plant Physiol. Biochem. 1999, 37, 789–794. [Google Scholar] [CrossRef]
- Wu, X.; Lin, J.; Lin, Q.; Wang, J.; Schreiber, L. Casparian strips in needles are more solute permeable than endodermal transport barriers in roots of Pinus bungeana. Plant Cell Physiol. 2005, 46, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Rioux, D.; Laflamme, G. Formation of ligno-suberized tissues in Jack pine resistant to the European race of Gremmeniella abietina. Phytopathology 2001, 91, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Brundrett, M.C.; Enstone, D.E.; Peterson, C.A. Berberine-aniline blue fluorescent staining procedure for suberin, lignin, and callose in plant tissue. Protoplasma 1988, 146, 133–142. [Google Scholar] [CrossRef]
- Shaw, S.L.; Ehrhardt, D.W. Smaller, faster, brighter: Advances in optical imaging of living plant cells. Annu. Rev. Plant Biol. 2013, 64, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006; p. 954. ISBN 0-387-31278-1. [Google Scholar]
- Frank, J.H.; Elder, A.D.; Swartling, J.; Venkitaraman, A.R.; Jeyasekharan, A.D.; Kaminski, C.F. A white light confocal microscope for spectrally resolved multidimensional imaging. J. Microsc. 2007, 227, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed]
- Broess, K.; Borst, J.W.; van Amerongen, H. Applying two-photon excitation fluorescence lifetime imaging microscopy to study photosynthesis in plant leaves. Photosynth. Res. 2009, 100, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Radotić, K. Fluorescence lifetime imaging of lignin autofluorescence in normal and compression wood. J. Microsc. 2013, 251, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Murchie, E.H.; Lawson, T. Chlorophyll fluorescence analysis: A guide to good practice and understanding some new applications. J. Exp. Bot. 2013, 64, 3983–3998. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Radotić, K.; Kalauzi, A.; Djikanović, D.; Jeremić, M. Quantification of compression wood severity in tracheids of Pinus radiata D. Don using confocal fluorescence imaging and spectral deconvolution. J. Struct. Biol. 2010, 169, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Knox, J.P. Localization of cell wall polysaccharides in normal and compression wood of radiata pine—Relationships with lignification and microfibril orientation. Plant Physiol. 2012, 158, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Kroese, H.W.; Hill, S.J.; Franich, R.A. Detection of wood cell wall porosity using small carbohydrate molecules and confocal fluorescence microscopy. J. Microsc. 2015, 259, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Vaidya, A. Visualizing recalcitrance by colocalization of cellulase, lignin and cellulose in pretreated pine biomass using fluorescence microscopy. Sci. Rep. 2017, 7, 44386. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, B.; Li, S.; Lundquist, K.; Stomberg, R. The origin of lignin fluorescence. J. Mol. Struct. 1999, 508, 19–27. [Google Scholar] [CrossRef]
- Donaldson, L.A. Softwood and hardwood lignin fluorescence spectra of wood cell walls in different mounting media. IAWA J. 2013, 34, 3–19. [Google Scholar] [CrossRef]
- Harris, P.J.; Hartley, R.D. Detection of bound ferulic acid in cell walls of the Gramineae by ultraviolet fluorescence microscopy. Nature 1976, 259, 508–510. [Google Scholar] [CrossRef]
- Wagner, A.; Donaldson, L.; Ralph, J. Lignification and lignin manipulations in conifers. Adv. Bot. Res. 2012, 61, 37–76. [Google Scholar] [CrossRef]
- Bernards, M.A. Demystifying suberin. Can. J. Bot. 2002, 80, 227–240. [Google Scholar] [CrossRef]
- Pollard, M.; Beisson, F.; Li, Y.; Ohlrogge, J.B. Building lipid barriers: Biosynthesis of cutin and suberin. Trends Plant Sci. 2008, 13, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Fich, E.A.; Segerson, N.A.; Rose, J.K.C. The plant polyester cutin: Biosynthesis, structure and biological roles. Annu. Rev. Plant Biol. 2016, 67, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, J.-P.; Jungblut, T.P.; Heller, W.; Köfferlein, M.; Hutzler, P.; Heinzmann, U.; Schmelzer, E.; Ernst, D.; Langebartels, C.; Sandermann, H. Tissue localization of UV-B screening pigments and of chalcone synthase mRNA in needles of Scots pine seedlings. New Phytol. 1996, 132, 247–258. [Google Scholar] [CrossRef]
- Strack, D.; Heilemann, J.; Klinkott, E.-S. Cell wall-bound phenolics from Norway spruce (Picea abies) needles. Z. Naturforsch. 1988, 43, 37–41. [Google Scholar] [CrossRef]
- Manninen, A.-M.; Tarhanen, S.; Vuorinen, M.; Kainulainen, P. Comparing the variation of needle and wood terpenoids in Scots pine provenances. J. Chem. Ecol. 2002, 28, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Pasqua, G.; Monacelli, B.; Manfredini, C.; Loreto, F.; Perez, G. The role of isoprenoid accumulation and oxidation in sealing wounded needles of Mediterranean pines. Plant Sci. 2002, 163, 355–359. [Google Scholar] [CrossRef]
- Pedros, R.; Moya, I.; Goulas, Y.; Jacquemoud, S. Chlorophyll fluorescence emission spectrum inside a leaf. Photochem. Photobiol. Sci. 2008, 7, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Dick, M.A.; Williams, N.M.; Bader, M.K.-F.; Gardner, J.F.; Bulman, L.S. Pathogenicity of Phytophthora pluvialis to Pinus radiata and its relation with red needle cast disease in New Zealand. N. Z. J. For. Sci. 2014, 44, 6. [Google Scholar] [CrossRef]
- Fester, T.; Berg, R.H.; Taylor, C.G. An easy method using glutaraldehyde-introduced fluorescence for the microscopic analysis of plant biotrophic interactions. J. Microsc. 2008, 231, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Jamme, F.; Kascakova, S.; Villette, S.; Allouche, F.; Pallu, S.; Rouam, V.; Refregliers, M. Deep UV autofluorescence microscopy for cell biology and tissue histology. Biol. Cell 2013, 106, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.A.; Nanayakkara, B.; Radotić, K.; Djikanovic-Golubovic, D.; Mitrovic, A.; Bogdanovic, J.; Simonovic, J.; Kalauzi, A. Xylem parenchyma cell walls lack a gravitropic response in conifer compression wood. Planta 2015, 242, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Meicenheimer, R.D.; Coffin, D.W.; Chapman, E.M. Anatomical basis for biophysical differences between Pinus nigra and P. resinosa (Pinaceae) leaves. Am. J. Bot. 2008, 95, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, K.; Stone, J.; Cook, K.; Sniezko, R.A.; Kegley, A.; Schoettle, A.W. Are needle reactions in resistance to Cronartium ribicola a hypersensitivity response? In Proceedings of the Fourth International Workshop on the Genetics of Host-Parasite Interactions in Forestry, Eugene, OR, USA, 31 July–5 August 2011; pp. 368–371. [Google Scholar]
- Fraser, S.; Martin-Garcia, J.; Perry, A.; Kabir, M.S.; Owen, T.; Solla, A.; Brown, A.V.; Bulman, L.S.; Barnes, I.; Hale, M.D.; et al. A review of Pinaceae resistance mechanisms against needle and shoot pathogens with a focus on the Dothistroma-Pinus interaction. For. Pathol. 2016, 46, 453–471. [Google Scholar] [CrossRef]
- Carnachan, S.M.; Harris, P.J. Ferulic acid is bound to the primary cell walls of all gymnosperm families. Biochem. Syst. Ecol. 2000, 28, 865–879. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Martz, F.; Sutinen, M.-L.; Derome, K.; Wingsle, G.; Julkunen-Tiitto, R.; Turunen, M. Effects of ultraviolet (UV) exclusion on the seasonal concentration of photosynthetic and UV-screening pigments in Scots pine needles. Glob. Chang. Biol. 2007, 13, 252–265. [Google Scholar] [CrossRef]
- Puech, L.; Mehne-Jakobs, B. Histology of magnesium-deficient Norway spruce needles influenced by nitrogen source. Tree Physiol. 1997, 17, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Laakso, K.; Sullivan, J.H.; Huttunen, S. The effects of UV-B radiation on epidermal anatomy in loblolly pine (Pinus taeda L.) and Scots pine (Pinus sylvestris L.). Plant Cell Environ. 2000, 23, 461–472. [Google Scholar] [CrossRef]
- Kivimäenpää, M.; Riikonen, J.; Sutinen, S.; Holopainen, T. Cell structural changes in the mesophyll of Norway spruce needles by elevated ozone and elevated temperature in open-field exposure during cold acclimation. Tree Phys. 2014, 34, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M. A microscopical study of the structure of leaves of the genus Pinus. Trans. Proc. R. Soc. N. Z. 1934, 63, 517–569. [Google Scholar]
- Riding, R.T.; Aitken, J. Needle structure, and development of the stomatal complex in cotyledons, primary needles, and secondary needles of Pinus radiata D. Don. Bot. Gaz. 1982, 143, 52–62. [Google Scholar] [CrossRef]
- Bäck, J.; Huttunen, S.; Kristen, U. Carbohydrate distribution and cellular injuries in acid rain and cold-treated spruce needles. Trees 1993, 8, 75–84. [Google Scholar] [CrossRef]
- Lhotáková, Z.; Urban, O.; Dubánková, M.; Cvikrovác, M.; Tomášková, I.; Kubínová, L.; Zvárae, K.; Marek, M.V.; Albrechtová, J. The impact of long-term CO2 enrichment on sun and shade needles of Norway spruce (Picea abies): Photosynthetic performance, needle anatomy and phenolics accumulation. Plant Sci. 2012, 188–189, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.S.; Ganley, R.J.; Bradshaw, R.E. The hemibiotrophic lifestyle of the fungal pine pathogen Dothistroma septosporum. For. Pathol. 2015, 45, 190–202. [Google Scholar] [CrossRef]
- Moëll, M.; Donaldson, L.A. Comparison of segmentation methods for digital image analysis of confocal microscope images to measure tracheid cell dimensions. IAWA J. 2001, 22, 267–288. [Google Scholar] [CrossRef]
- Sokal, R.R.; Rohlf, F.J. Biometry; WH Freeman: San Francisco, CA, USA, 1981; p. 859. ISBN 0-7167-1254-7. [Google Scholar]
- Neher, R.A.; Mitkovski, M.; Kirchhoff, F.; Neher, E.; Theis, F.J.; Zeug, A. Blind source separation techniques for the decomposition of multiply labeled fluorescence images. Biophys. J. 2009, 96, 3791–3800. [Google Scholar] [CrossRef] [PubMed]
- Nakano, J.; Meshitsuka, G. The detection of lignin. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin, Germany, 1992; pp. 23–32. ISBN 3-540-50295-5. [Google Scholar]
- Biggs, A.R. Intracellular suberin: Occurrence and detection in tree bark. IAWA J. 1984, 5, 243–248. [Google Scholar] [CrossRef]
- David, R.; Carde, J.P. Coloration différentielle des inclusions lipidiques et therpéniques des pseudophylles du pin maritime an moyen du réactif Nadi. C. R. Acad. Sci. Paris 1964, 258, 1338–1340. [Google Scholar]
- Reeve, R.M. Histological and histochemical changes in developing and ripening peaches. III. Catechol tannin content per cell. Am. J. Bot. 1959, 46, 645–650. [Google Scholar] [CrossRef]
- Gardner, R.O. Vanillin-hydrochloric acid as a histochemical test for tannin. Stain Technol. 1975, 50, 315–317. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorophore | Excitation | Emission | λmax |
|---|---|---|---|
| Extractive (mesophyll) | 355 nm | Blue | 455 nm |
| Suberin | 355 nm | Blue | 465 nm |
| Lignin | 355/488 nm | Blue/Green | 455/535 nm |
| Ferulate | 355 nm | Green (pH9) | 480 nm |
| Flavonoid | 488 nm | Green | 535 nm |
| Chlorophyll | 355/488/561/633 nm | Red | 685/730 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donaldson, L.; Williams, N. Imaging and Spectroscopy of Natural Fluorophores in Pine Needles. Plants 2018, 7, 10. https://doi.org/10.3390/plants7010010
Donaldson L, Williams N. Imaging and Spectroscopy of Natural Fluorophores in Pine Needles. Plants. 2018; 7(1):10. https://doi.org/10.3390/plants7010010
Chicago/Turabian StyleDonaldson, Lloyd, and Nari Williams. 2018. "Imaging and Spectroscopy of Natural Fluorophores in Pine Needles" Plants 7, no. 1: 10. https://doi.org/10.3390/plants7010010
APA StyleDonaldson, L., & Williams, N. (2018). Imaging and Spectroscopy of Natural Fluorophores in Pine Needles. Plants, 7(1), 10. https://doi.org/10.3390/plants7010010