
Structural Properties of Cruciferin and Napin of Brassica napus (Canola) Show Distinct Responses to Changes in pH and Temperature


Abstract
:1. Introduction
2. Results
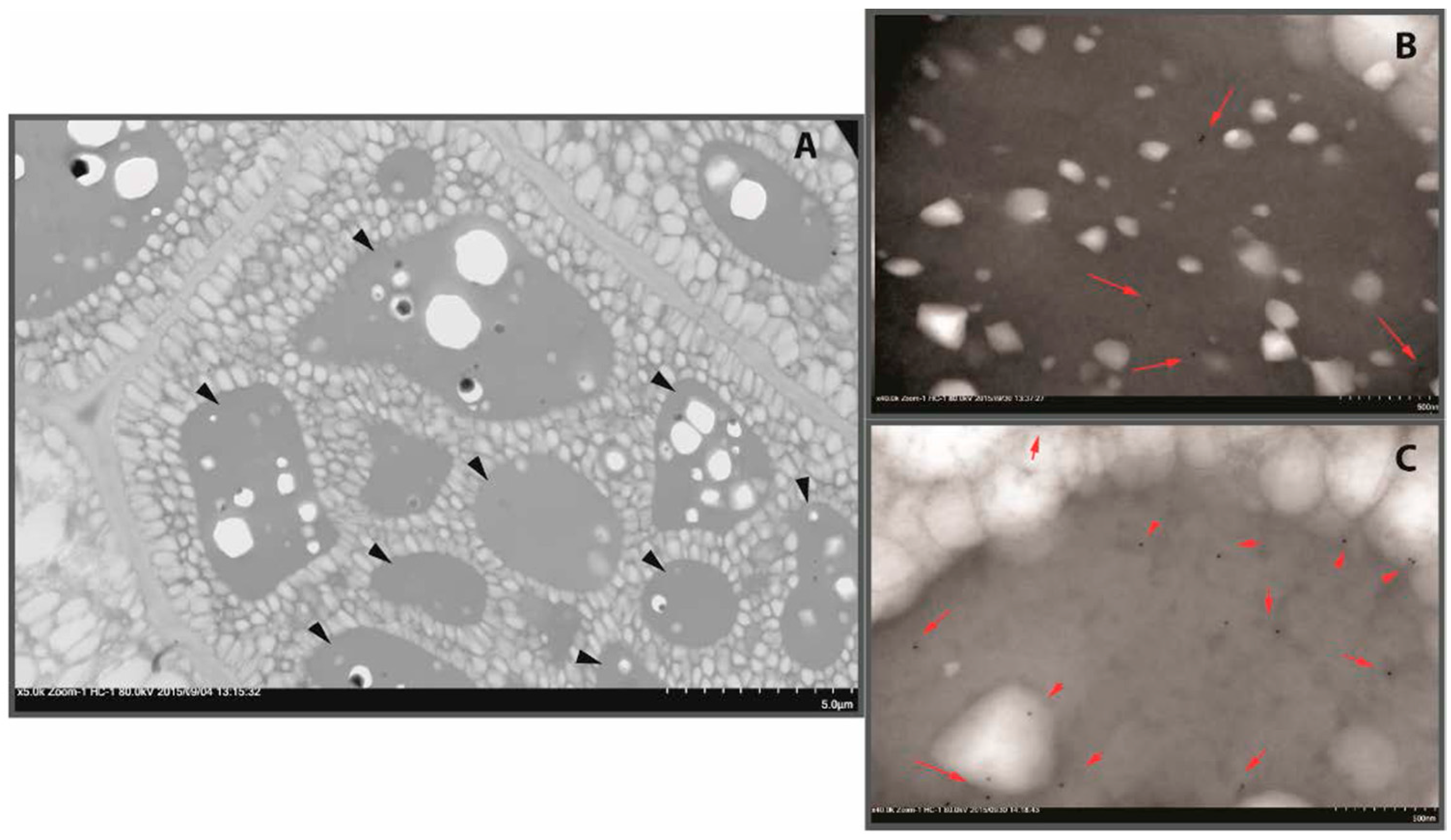
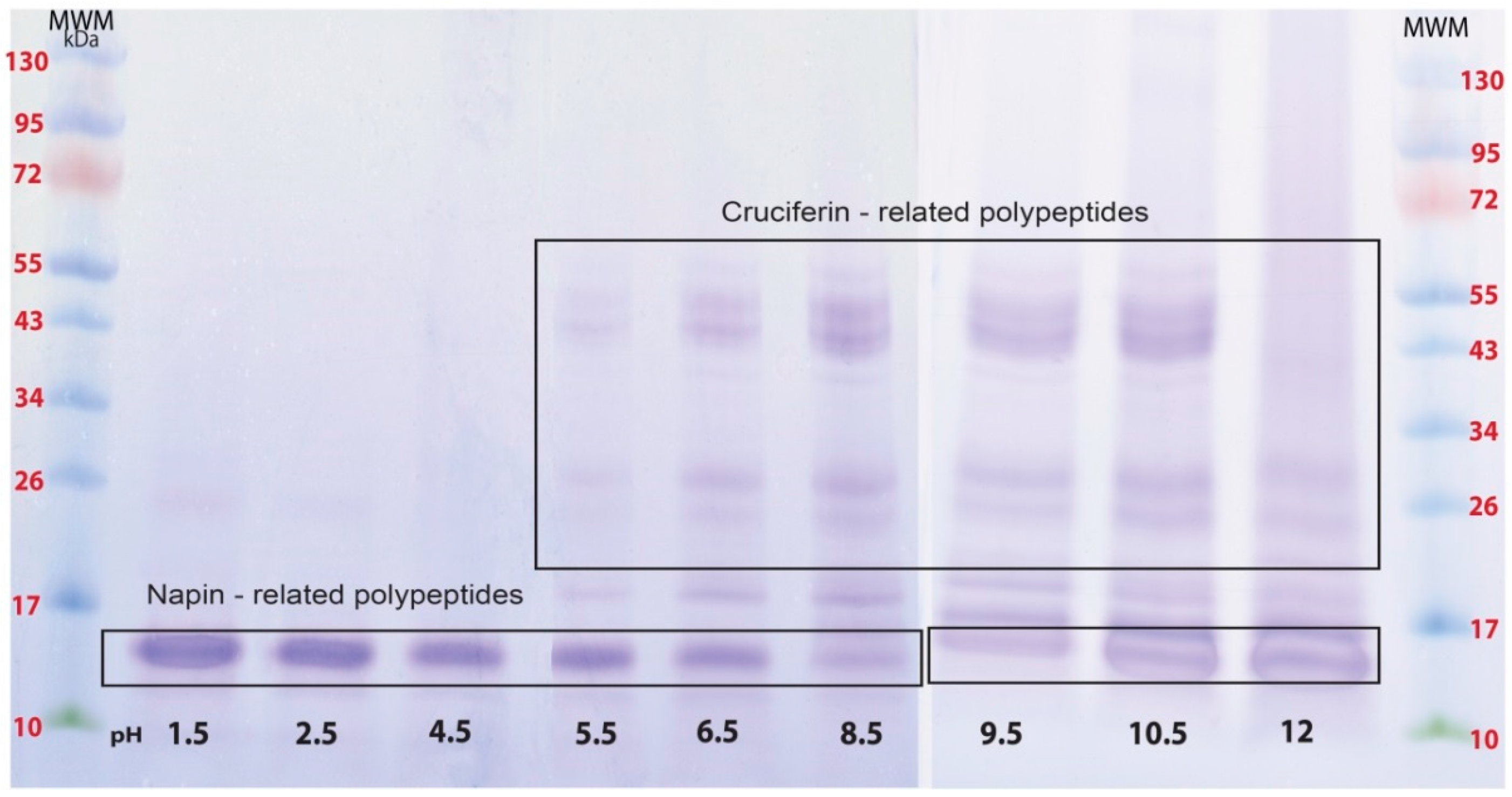
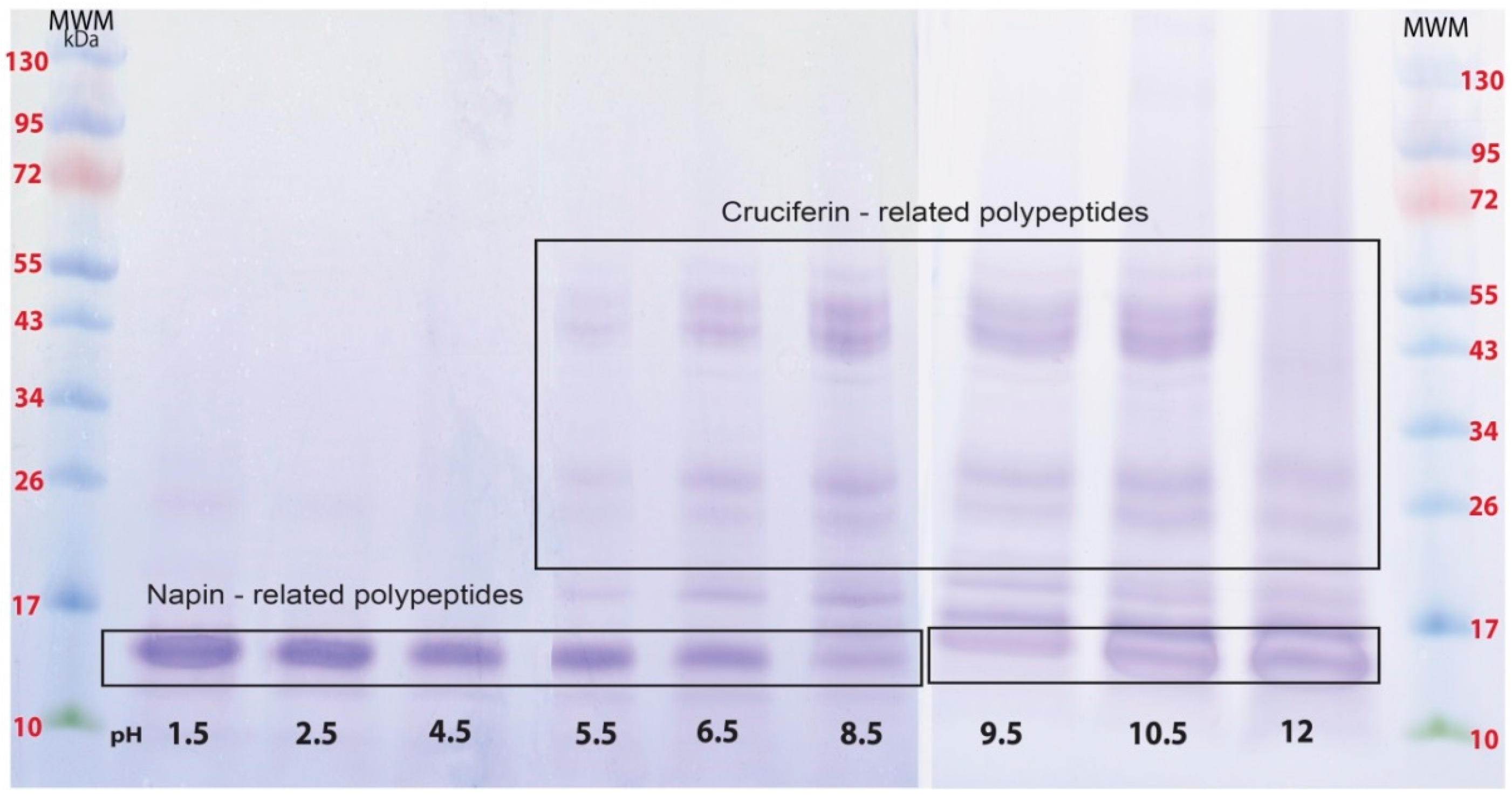
2.1. Proteins of B. napus Meal and Their Solubility Behaviour with pH Change
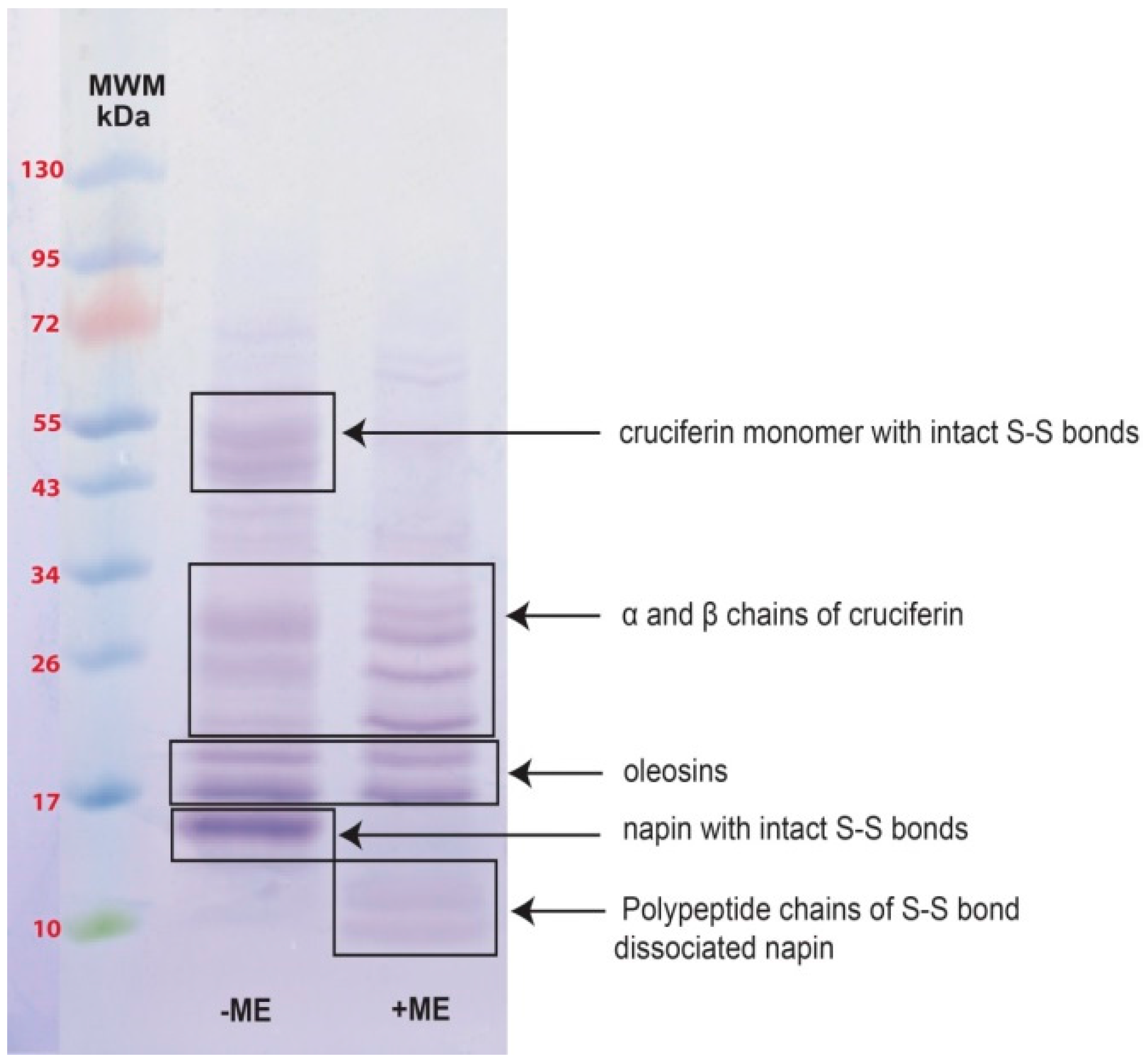
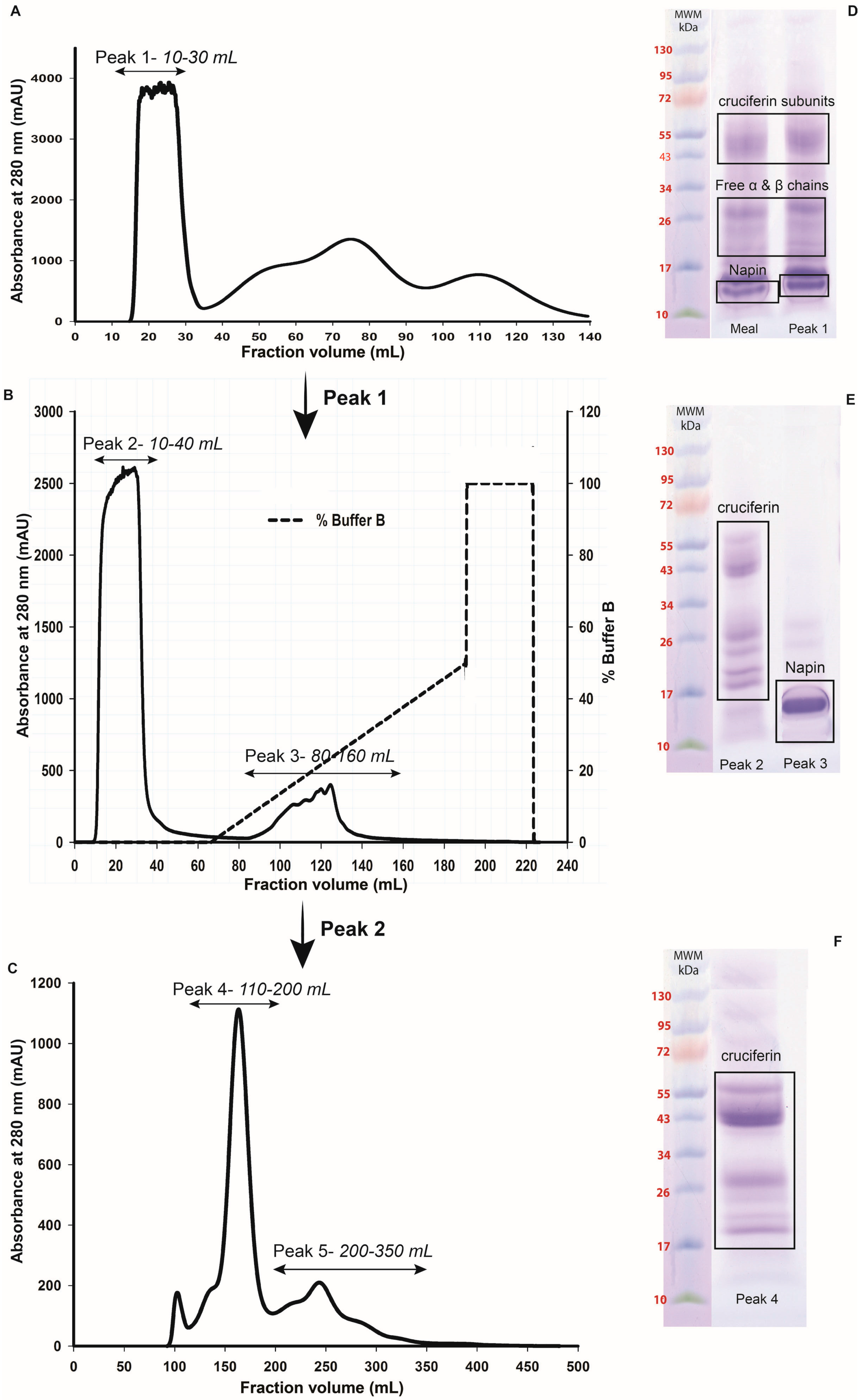
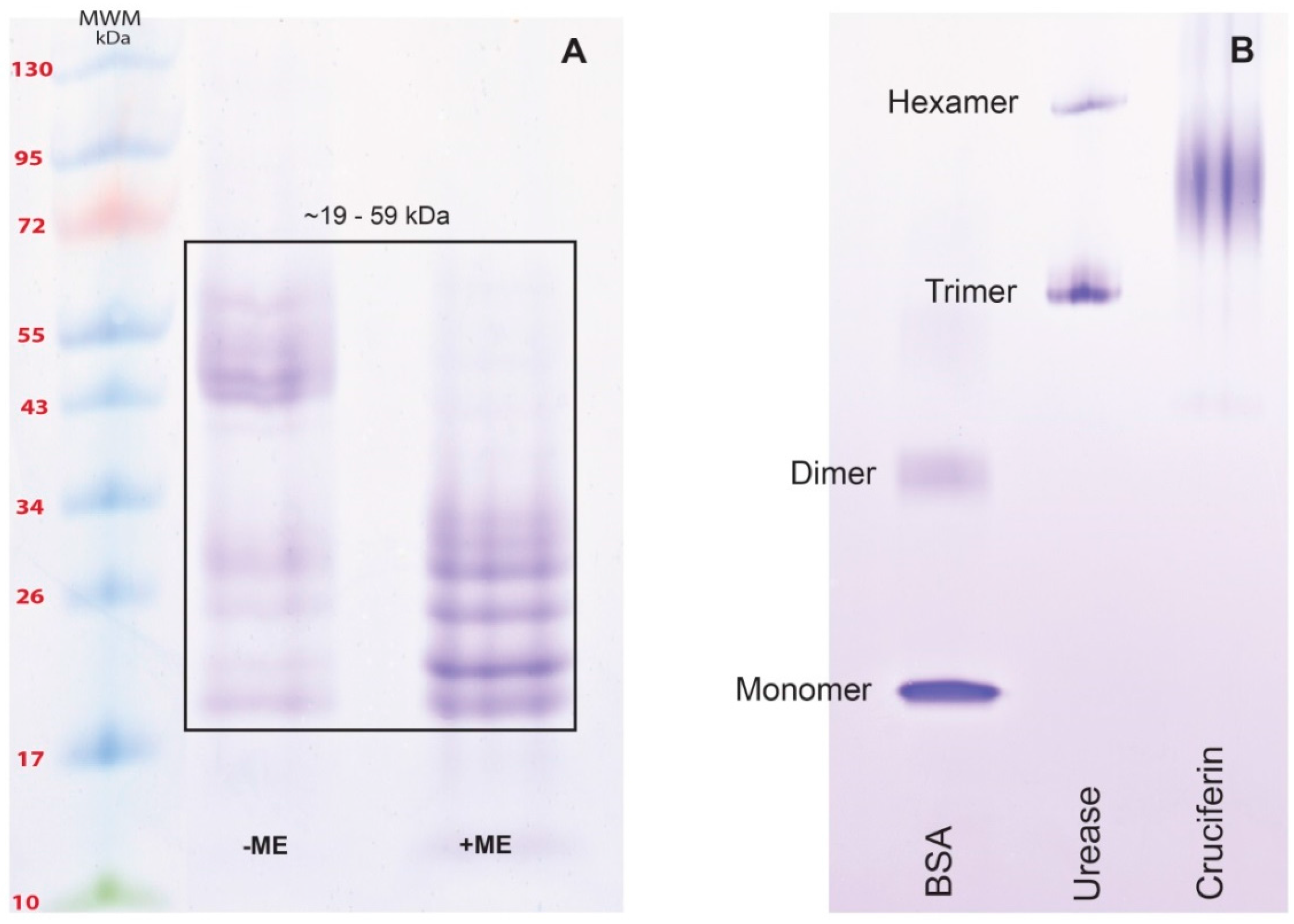
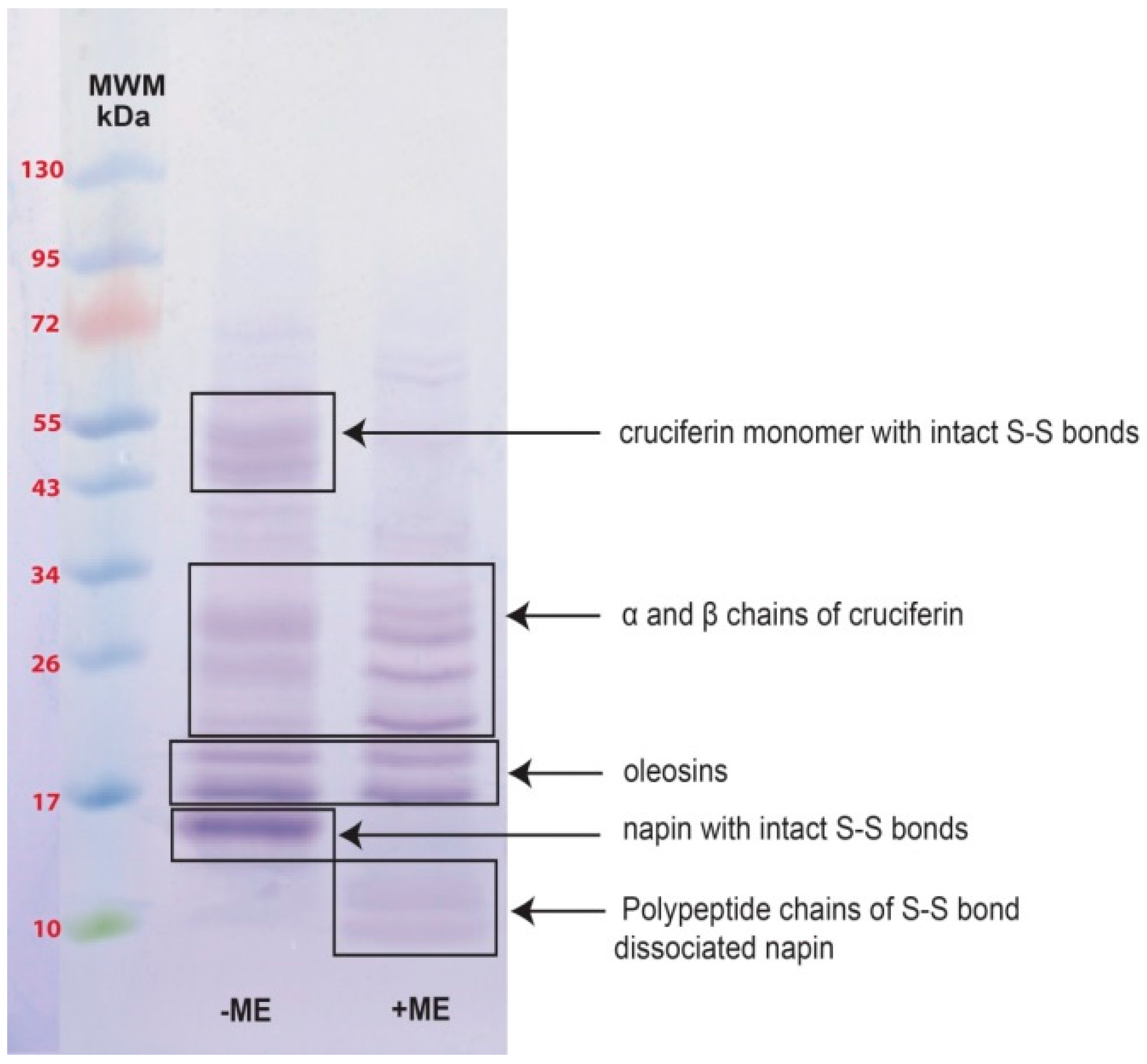
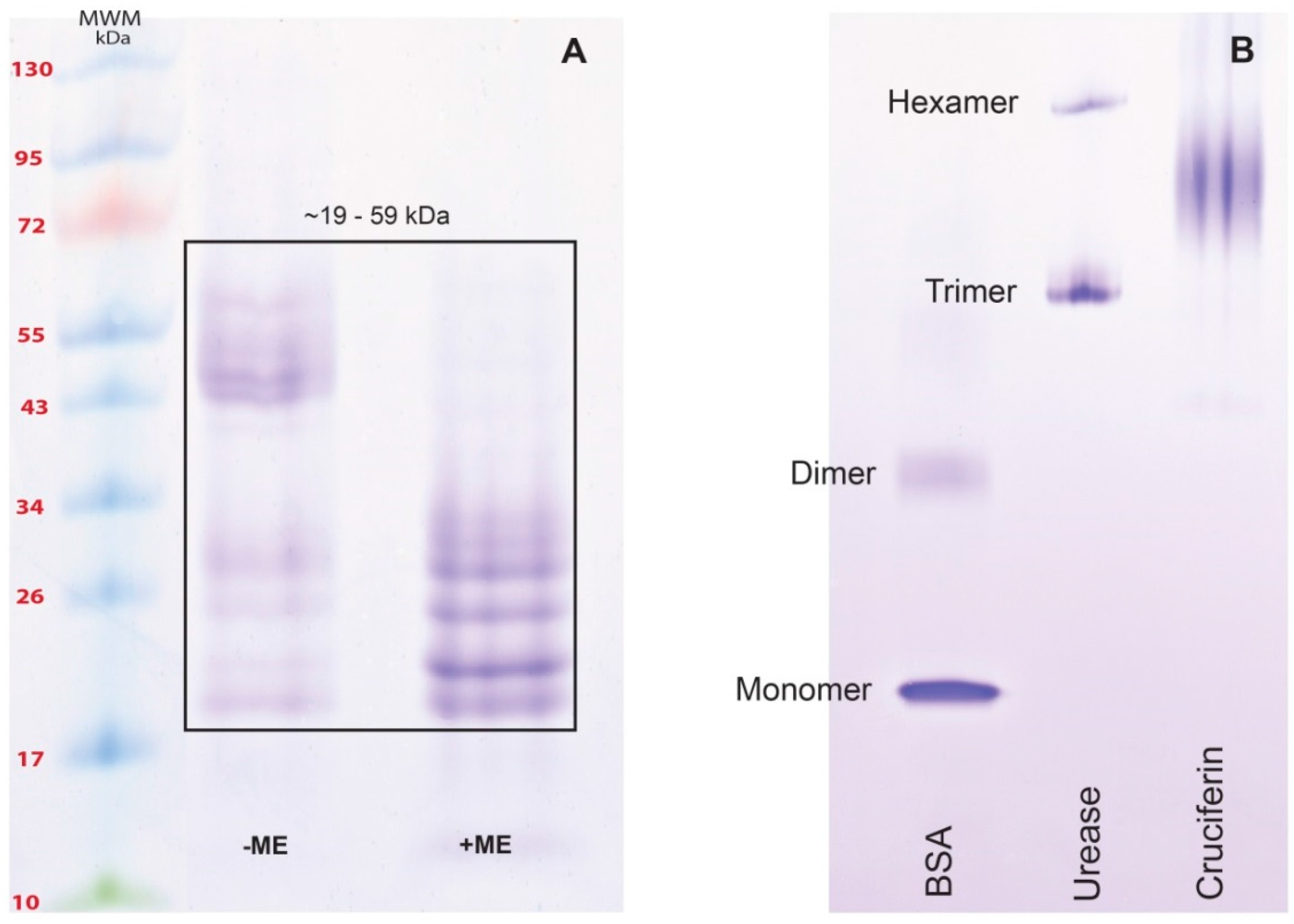
2.2. Obtaining Cruciferin
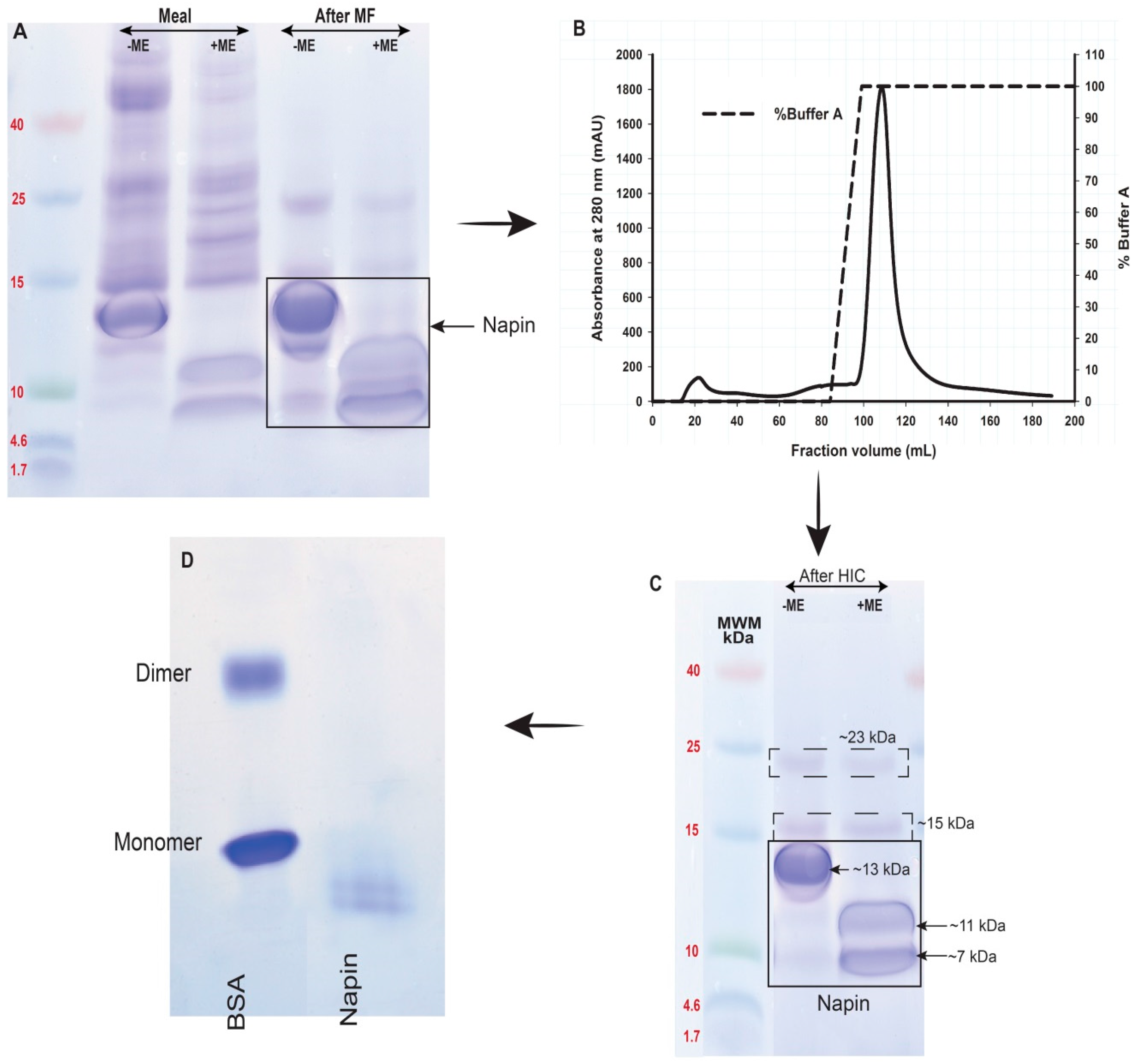
2.3. Obtaining Napin
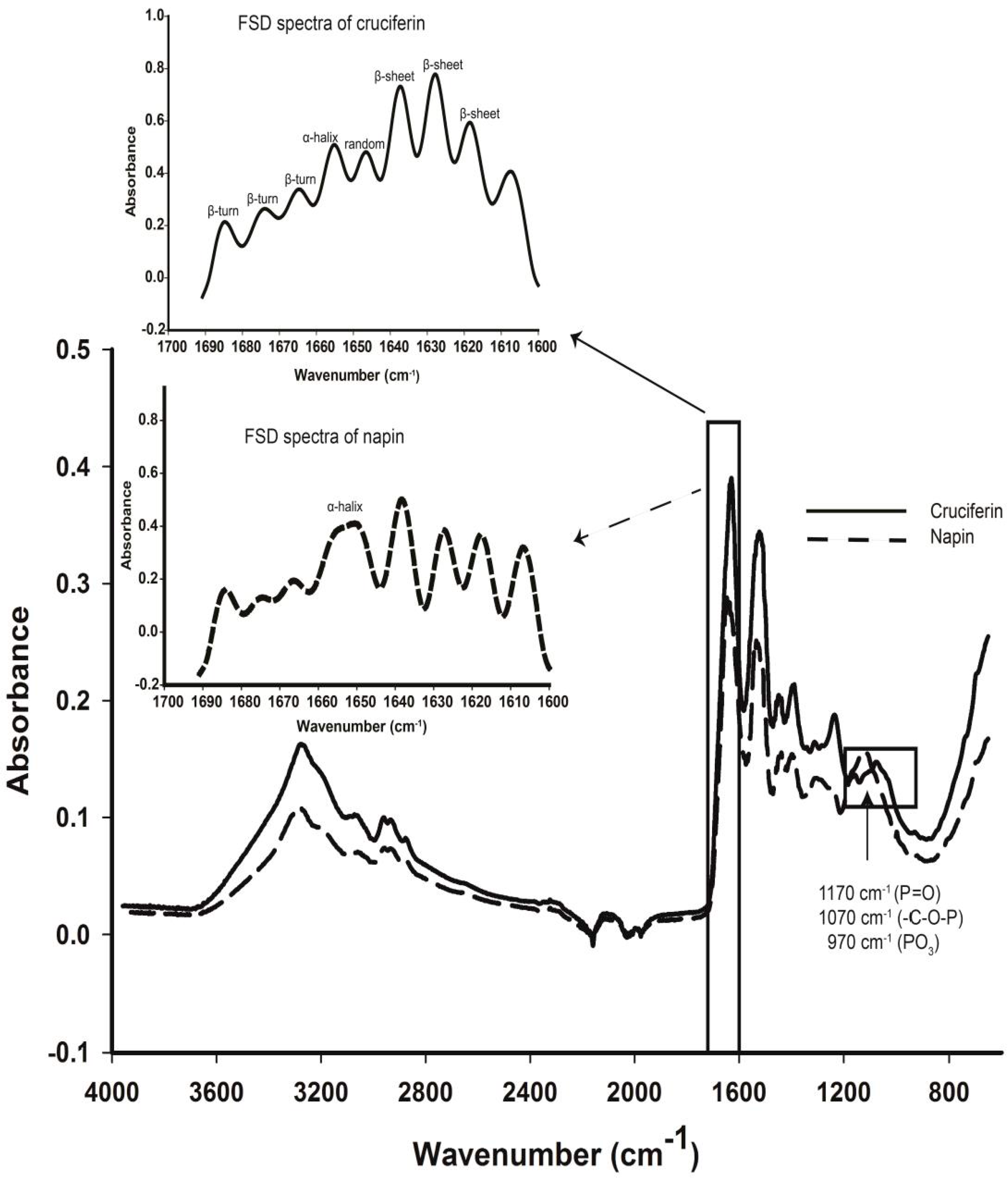
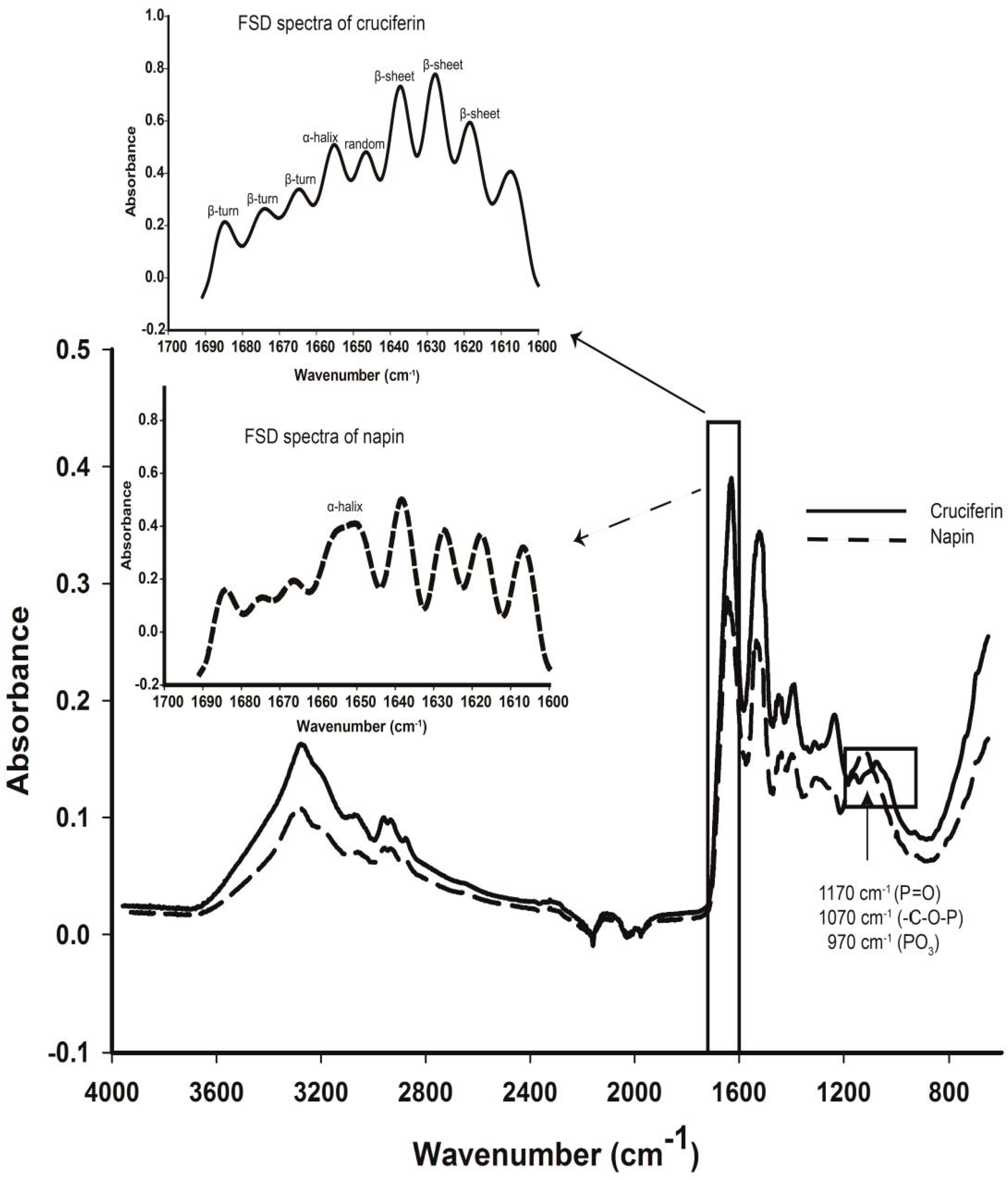
2.4. Secondary Structure Features of Cruciferin and Napin and Their Changes Due to Medium pH
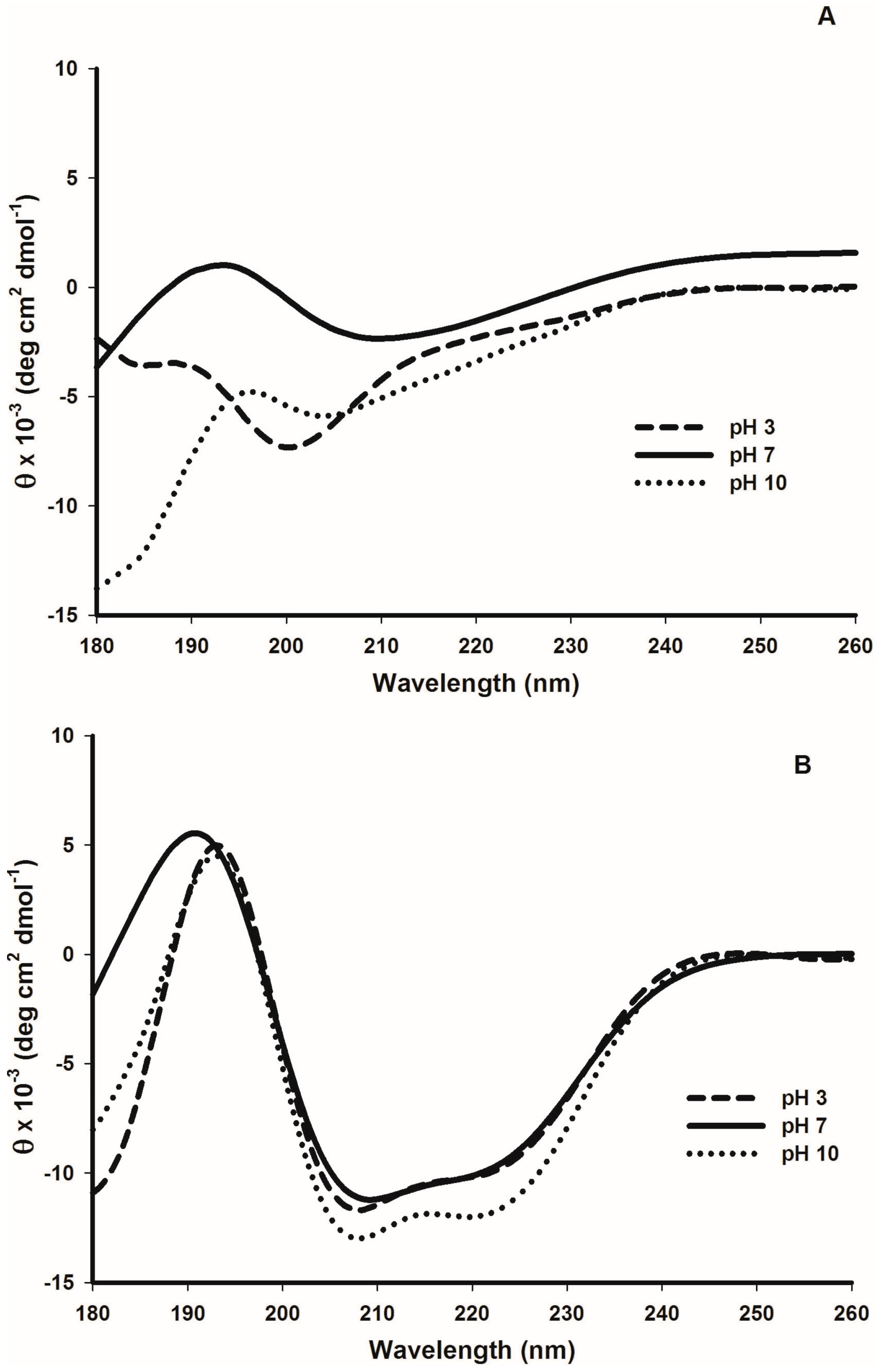
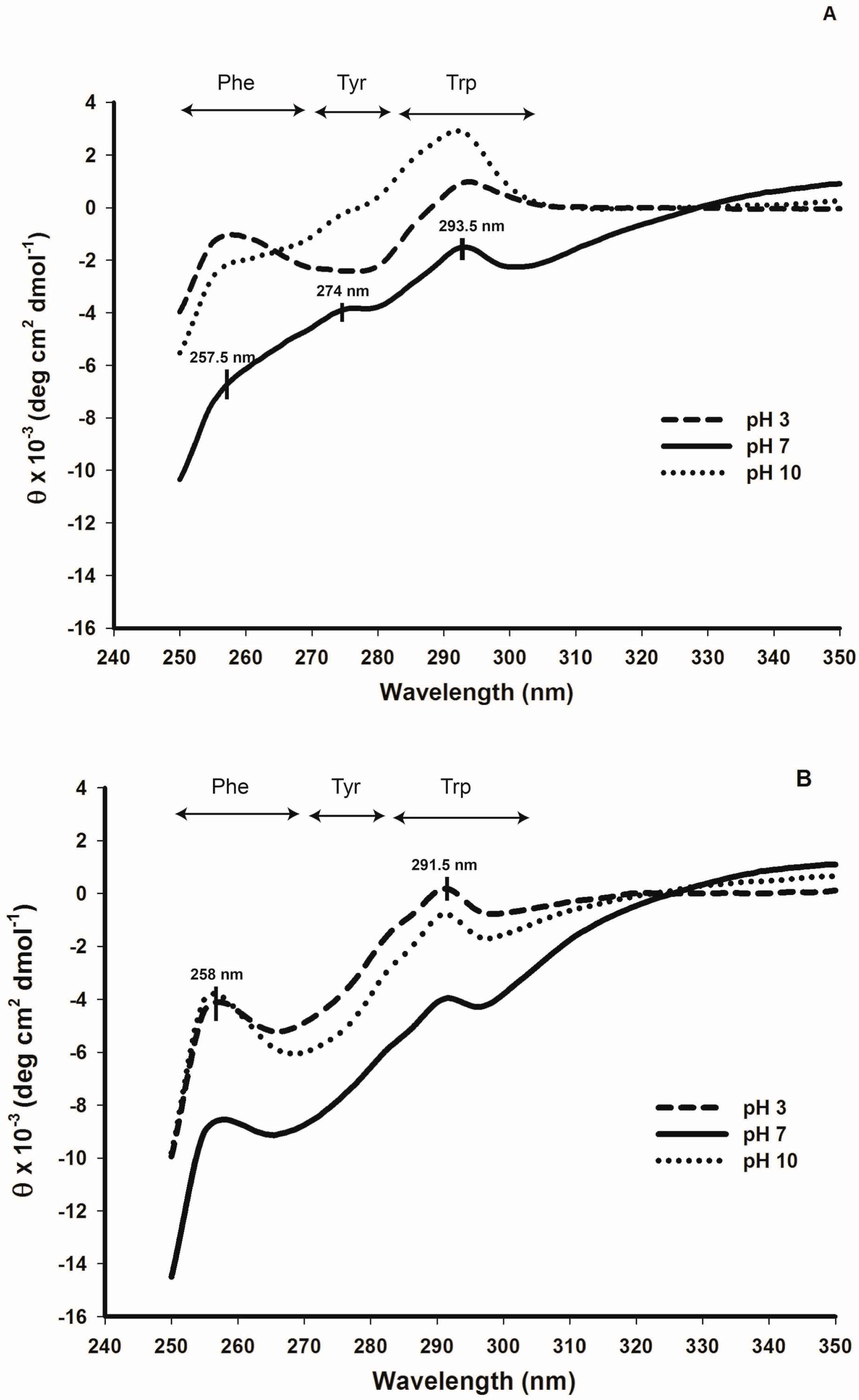
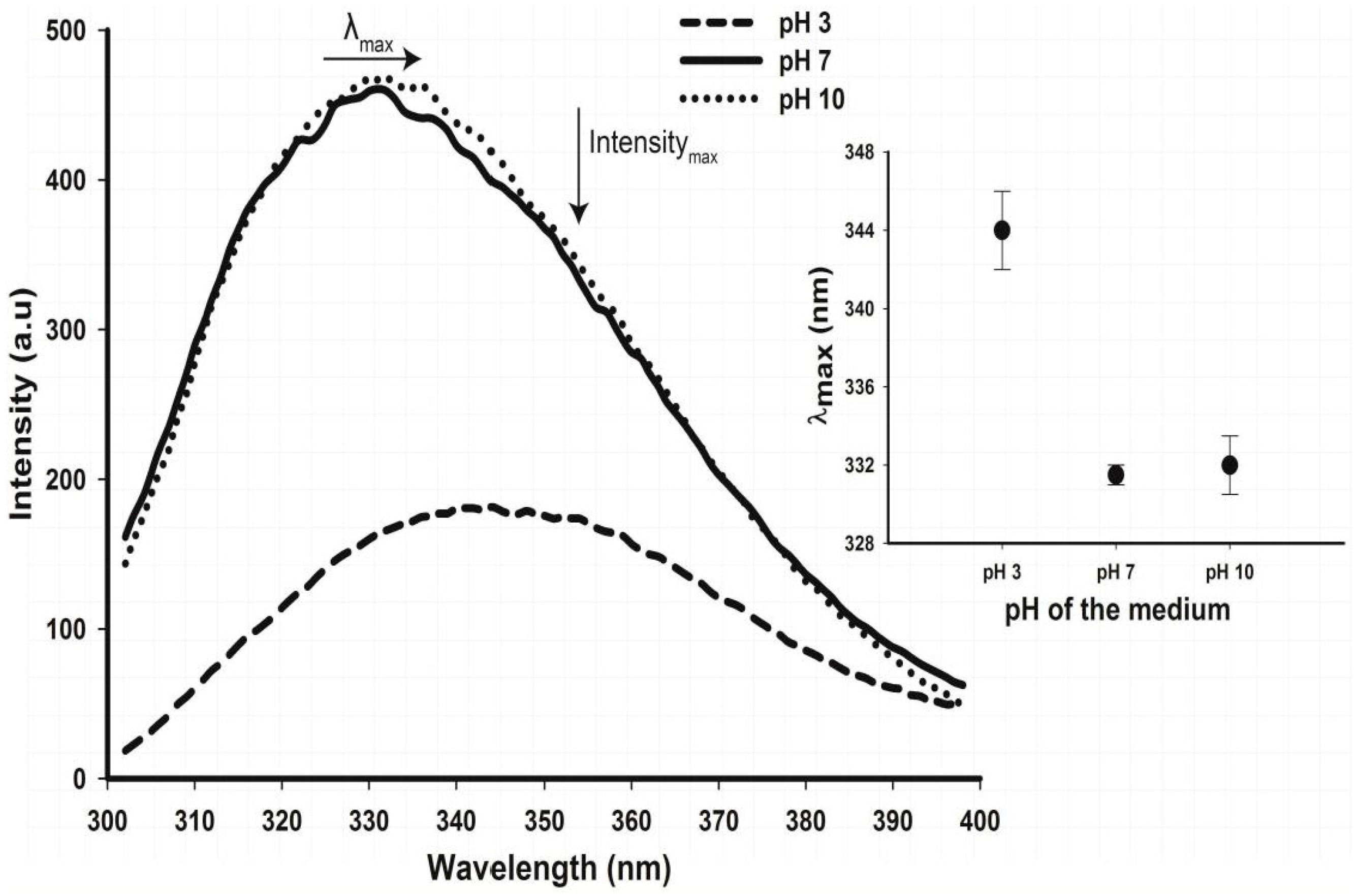
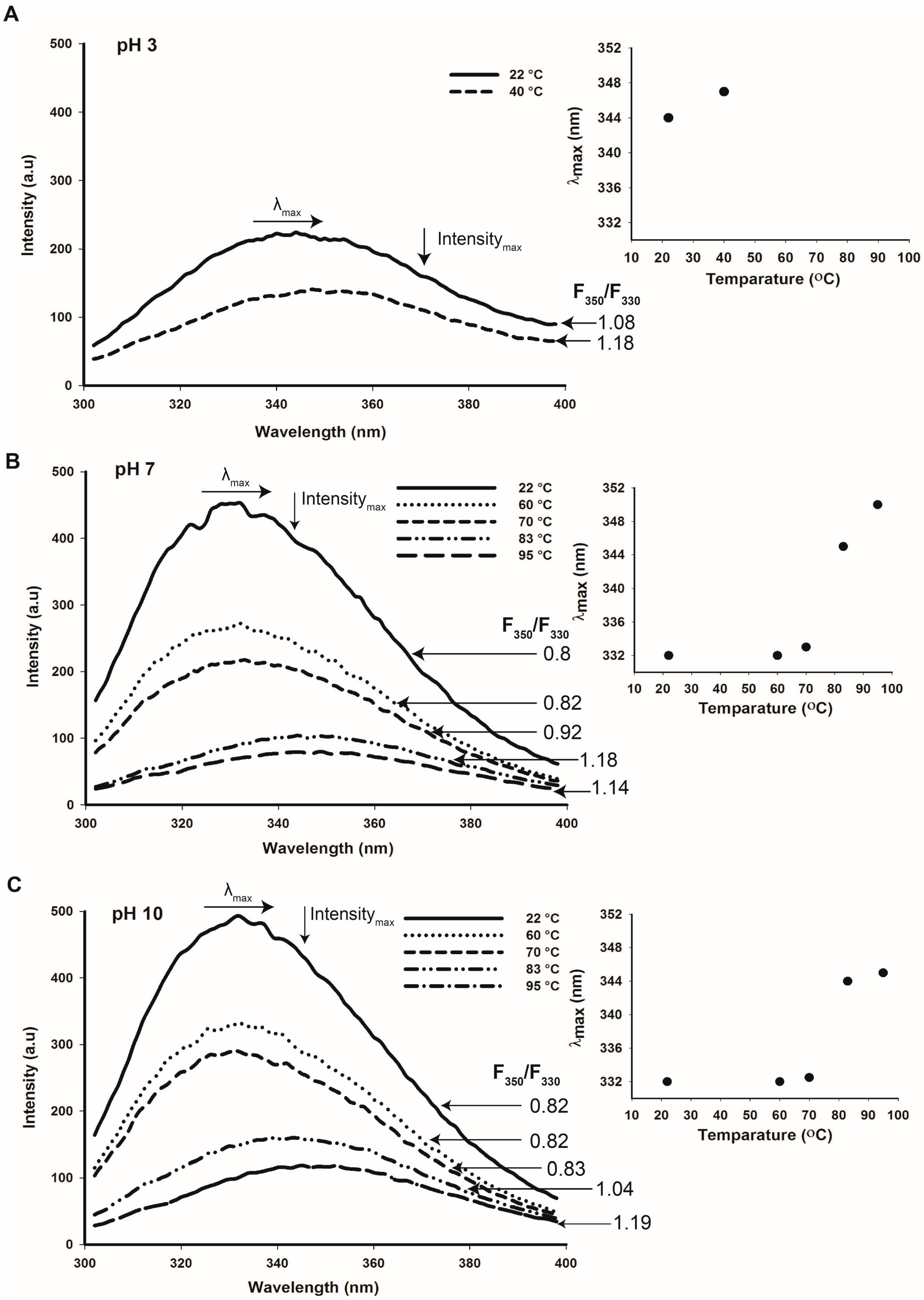
2.5. Tertiary Structure Details of Cruciferin and Napin and Their Changes with Medium pH
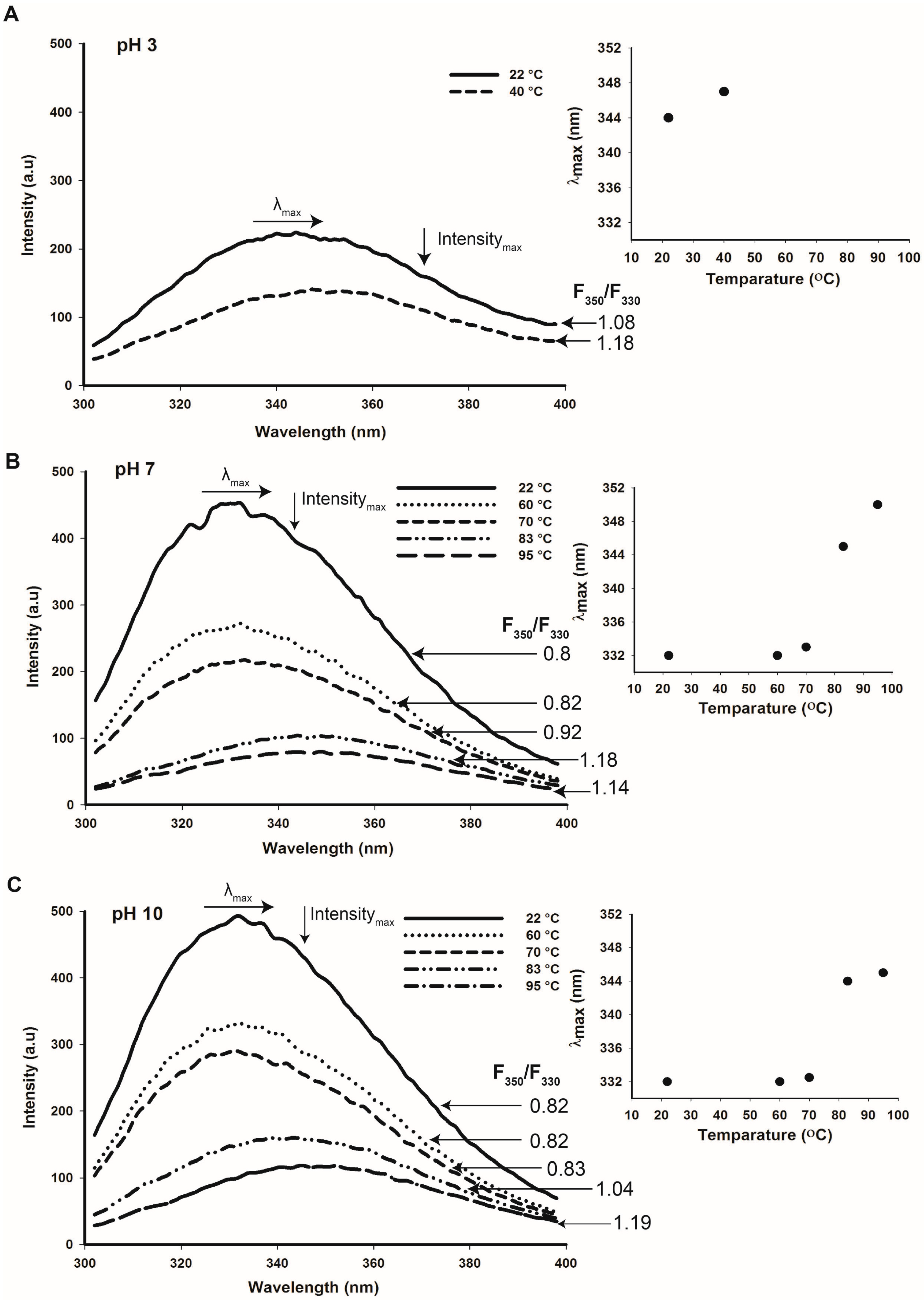
2.6. Thermal Stability of Cruciferin and Napin Structure at Different pHs
3. Discussion
3.1. Obtaining Cruciferin and Napin
3.2. Effect of pH and Temperature on Cruciferin
3.3. Effect of pH and Temperature on Napin
3.4. Relevance to Seed Protein Utilization
4. Materials and Methods
4.1. Seeds
4.2. Meal and Meal Protein
4.2.1. Preparation of Meal
4.2.2. Types of Protein Soluble at Different pHs
4.3. Preparation of Purified Cruciferin
4.3.1. Extracting Meal Protein
4.3.2. Separation and Purification of Cruciferin
4.4. Isolation and Purification of Napin at Low pH
4.5. N-Based Protein Content
4.6. Sodium Dodecyl Sulfite Polyacrylamide Gel Electrophoresis (SDS-PAGE)
4.7. Native Polyacrylamide Gel Electrophoresis (Native-PAGE)
4.8. FT-IR Spectroscopy
4.9. Circular Dichroism (CD) Spectroscopy
4.10. Fluorescence Spectroscopy
4.10.1. Intrinsic Fluorescence
4.10.2. Surface Hydrophobicity
4.11. Differential Scanning Calorimetry (DSC)
4.12. Experimental Design and Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Crouch, M.L.; Sussex, I.M. Development and storage-protein synthesis in Brassica napus L. embryos in vivo and in vitro. Planta 1981, 153, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Höglund, A.-S.; Rödin, J.; Larsson, E.; Rask, L. Distribution of napin and cruciferin in developing rape seed embryos. Plant Physiol. 1992, 98, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, I.; Renard, D.; Rondeau, D.; Richomme, P.; Popineau, Y.; Axelos, M.A. Detailed physicochemical characterization of the 2S storage protein from rape (Brassica napus L.). J. Agric. Food Chem. 2004, 52, 5995–6001. [Google Scholar] [CrossRef] [PubMed]
- Nietzel, T.; Dudkina, N.V.; Haase, C.; Denolf, P.; Semchonok, D.A.; Boekema, E.J.; Braun, H.P.; Sunderhaus, S. The native structure and composition of the cruciferin complex in Brassica napus. J. Biol. Chem. 2013, 288, 2238–2245. [Google Scholar] [CrossRef] [PubMed]
- Wanasundara, J.P.D. Proteins of Brassicaceae oilseeds and their potential as a plant protein source. Crit. Rev. Food Sci. Nutr. 2011, 51, 635–677. [Google Scholar] [CrossRef] [PubMed]
- Bérot, S.; Compoint, J.P.; Larré, C.; Malabat, C.; Guéguen, J. Large scale purification of rapeseed proteins (Brassica napus L.). J. Chromatogr. 2005, 818, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Shewry, P.R. Seed proteins. In Seed Technology and Its Biological Basis; Black, M., Bewley, J., Eds.; CRC Press: Boca Raton, FL, USA, 2000; pp. 42–84. [Google Scholar]
- Shewry, P.R.; Napier, J.A.; Tatham, A.S. Seed storage proteins: Structures and biosynthesis. Plant Cell 1995, 7, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Schwenke, K.D.; Raab, B.; Plietz, P.; Damaschun, G. The structure of the 12S globulin from rapeseed. Nahrung 1983, 27, 165–175. [Google Scholar] [CrossRef]
- Tandang-Silvas, M.R.G.; Fukuda, T.; Fukuda, C.; Prak, K.; Cabanos, C.; Kimura, A.; Itohb, T.; Mikamic, B.; Utsumi, S.; Maruyama, N. Conservation and divergence on plant seed 11S globulins based on crystal structures. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Sjödahl, S.; Rödin, J.; Rask, L. Characterization of the 12S globulin complex of Brassica napus. Evolutionary relationship to other 11-12S storage globulins. Eur. J. Biochem. 1991, 196, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Withana-Gamage, T.S. Structure and Properties of Cruciferin: Investigation of Homohexameric Cruciferin Expressed in Arabidopsis. Ph.D. Thesis, University of Saskatchewan, Saskatoon, SK, Canada, 2013. [Google Scholar]
- Gruis, D.F.; Selinger, D.A.; Curran, J.M.; Jung, R. Redundant proteolytic mechanisms process seed storage proteins in the absence of seed-type members of the vacuolar processing enzyme family of cysteine proteases. Plant Cell 2000, 14, 2863–2882. [Google Scholar] [CrossRef]
- Otegui, M.S.; Herder, R.; Schulze, J.; Jung, R.; Staehelin, L.A. The Proteolytic Processing of Seed Storage Proteins in Arabidopsis Embryo Cells Starts in the Multivesicular Bodies. Plant Cell 2006, 18, 2567–2581. [Google Scholar] [CrossRef] [PubMed]
- Scofield, R.; Crouch, M.J. Nucleotide sequence of a member of the napin storage protein family from Brassica napus. J. Biol. Chem. 1987, 262, 12202–12208. [Google Scholar] [PubMed]
- Raynal, M.; Depigny, D.; Grellet, F.; Deslseny, M. Characterization and evolution of napin encoding genes in radish and related crucifers. Gene 1991, 99, 77–86. [Google Scholar] [CrossRef]
- UniPort. Available online: http://www.uniprot.org/ (accessed on 8 November 2015).
- Rico, M.; Bruix, M.; Gonzalez, C.; Monsalve, R.I.; Rodriguez, R. 1H NMR assignment and global fold of napin BnIb, a representative 2S albumin seed protein. Biochemistry 1996, 35, 15672–15682. [Google Scholar] [CrossRef] [PubMed]
- Apenten, R.K.O.; Folawiyo, Y.L. The effect of pH on rapeseed globulin (cruciferin) binding to anilinonaphthalene sulfonate. J. Food Biochem. 1995, 19, 455–465. [Google Scholar] [CrossRef]
- Wijesundera, C.; Boiteau, T.; Xu, X.; Shen, Z.; Watkins, P.; Logan, A. Stabilization of fish oil-in-water emulsions with oleosin extracted from canola meal. J. Food Sci. 2013, 78, C1340–C1347. [Google Scholar] [CrossRef] [PubMed]
- Matulis, D.; Baumann, C.G.; Bloomfield, V.A.; Lovrien, R.E. 1-Anilino-8-naphthalene sulfonate as a protein conformational tightening agent. Biopolymers 1999, 49, 451–458. [Google Scholar] [CrossRef]
- Fluorescence Data Analysis Methods. Available online: http://www.isogen-lifescience.com/uploads/o4/21/o4210uzD8OIHHtyOEhZEkQ/Optim06v1.0---Fluorescence-data-analysis-methods-ILS.pdf (accessed on 30 August 2016).
- Tan, S.H.; Mailer, R.J.; Blanchard, C.L.; Agboola, S.O. Canola proteins for human consumption: Extraction, profile, and functional properties. J. Food Sci. 2011, 76, R16–R28. [Google Scholar] [CrossRef] [PubMed]
- Aluko, R.E.; McIntosh, T. Polypeptide profile and functional properties of defatted meals and protein isolates of canola seeds. J. Food Sci. Agric. 2001, 81, 391–396. [Google Scholar] [CrossRef]
- Inquello, V.; Raymond, J.; Azanza, J.L. Disulfide interchange reactions in 11S globulin subunits of Cruciferae seeds. Eur. J. Biochem. 1993, 217, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Jyothi, T.C.; Sinha, S.; Singh, S.A.; Surolia, A.; Appu Rao, A.G. Napin from Brassica juncea: Thermodynamic and structural analysis of stability. Biochim. Biophys. Acta 2007, 1774, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, R.I.; Lopez-Otin, C.; Villalba, M.; Rodríguez, R. A new distinct group of 2S albumins from rapeseed: Amino acid sequence of two low molecular weight napins. FEBS Lett. 1991, 295, 207–210. [Google Scholar] [CrossRef]
- Wanasundara, J.P.D.; Abeysekara, S.J.; McIntosh, T.C.; Falk, K.C. Solubility differences of major storage proteins of Brassicaceae oilseeds. J. Am. Oil Chem. Soc. 2012, 89, 869–881. [Google Scholar] [CrossRef]
- Wanasundara, J.P.D.; McIntosh, T.C. Process of aqueous protein extraction from Brassicaceae oilseeds. Available online: http://www.google.com/patents/US8557963 (accessed on 2 January 2016).
- Dalgalarrondo, M.; Robin, J.-M.; Azanza, J.-L. Subunit composition of the globulin fraction of rapeseed (Brassica napus L.). Plant Sci. 1986, 43, 115–124. [Google Scholar] [CrossRef]
- Job, C.; Rajjou, L.; Lovigny, Y.; Belghazi, M.; Job, D. Patterns of protein oxidation in Arabidopsis seeds and during germination. Plant Physiol. 2005, 138, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.; Nam, Y.W.; Saalbach, I.; Muntz, K.; Nielsen, N.C. Role of the sulfhydryl redox state and disulfide bonds in processing and assembly of 11S seed globulins. Plant Cell 1997, 9, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Kanamori, J.; Masuda, T.; Yagasaki, K.; Kitamura, K.; Mikami, B.; Utsumi, S. Crystal structure of soybean 11S globulin: Glycinin A3B4 homohexamer. Proc. Natl. Acad. Sci. USA 2003, 100, 7395–7400. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Winkler, U.; Stabenau, H. Separation of Native Basic Protein by Cathodic, Discontinuous Polyacrylamide Gel Electrophoresis. Available online: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_2376.pdf (accessed on 10 February 2016).
- Chen, Y.-C.; Liu, W.-L.; Hsu, H.-C.; Lee, Y.A.; Chen, C.-S. Purification and characterization of isoforms of b-N-acetylhexosaminidase from mungbean seedlings. Bot. Bull. Acad. Sin. 2004, 45, 275–283. [Google Scholar]
- Fink, A.L.; Seshadri, S.; Khurana, R.; Oberg, K.A. Determination of secondary structure in protein aggregates using attenuated total reflectance FTIR infrared. Anal. Pept. Proteins 1999, 750, 132–144. [Google Scholar]
- Shivu, B.; Seshadri, S.; Li, J.; Oberg, K.A.; Uversky, V.N.; Fink, A.L. Distinct beta-sheet structure in protein aggregates determined by ATR-FTIR spectroscopy. Biochemistry 2013, 52, 5176–5183. [Google Scholar] [CrossRef] [PubMed]
- Stryer, L. Fluorescence Spectroscopy of Proteins. Science 1968, 162, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Slavik, J. Anilinonaphthalene sulfonate as a probe of membrane composition and function. Biochim. Biophys. Acta 1982, 694, 1–25. [Google Scholar] [CrossRef]
- Arakawa, T.; Kita, Y.A.; Narhi, L.O. Protein-ligand interaction as a method to study surface properties of proteins. Methods Biochem. Anal. 1991, 35, 87–125. [Google Scholar] [PubMed]
- Korte, T.; Herrmann, A. pH-dependent binding of the fluorophore bis-ANS to influenza virus reflects the conformational change of hemagglutinin. Eur. Biophys. J. 1994, 23, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lakemond, C.M.; de Jongh, H.H.; Hessing, M.; Gruppen, H.; Voragen, A.G. Heat denaturation of soy glycinin: Influence of pH and ionic strength on molecular structure. J. Agric. Food Chem. 2000, 48, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Weichel, M.; Bassarab, S.; Garidel, P. Probing Thermal Stability of MAbs by Intrinsic Tryptophan Fluorescence. Available online: http://www.bioprocessintl.com/manufacturing/antibody-non-antibody/probing-thermal-stability-of-mabs-by-intrinsic-tryptophan-fluorescence-182648/ (accessed on 5 January 2016).
- Eftink, M.R. The use of fluorescence methods to monitor unfolding transition in protein. Biophys. J. 1994, 66, 482–501. [Google Scholar] [CrossRef]
- Fink, A.L. Molten Globule. In eLS; John Wiley & Sons, Ltd.: Malden, MA, USA, 2001. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, S.; Yang, H.J.; Kwon, D.Y. Changes of glycinin conformation due to pH, heat and salt determined by differential scanning calorimetry and circular dichroism. Int. J. Food Sci. Tech. 2004, 39, 385–393. [Google Scholar] [CrossRef]
- Bhatty, R.S.; McKenzie, S.L.; Finlayson, A.J. The proteins of rapeseed (Brassica napus L.) soluble in salt solutions. Can. J. Biochem. 1968, 46, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Schwenke, K.D.; Linow, K.J. A reversible dissociation of the 12S globulin from rapeseed (Brassica napus L.) depending on ionic strength. Food/Nahrung 1982, 26, K5–K6. [Google Scholar] [CrossRef]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.L.; Lipscomb, W.N.; Schellman, C.G. The reverse turn as a polypeptide conformation in globular proteins. Proc. Natl. Acad. Sci. USA 1973, 70, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, C.M. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 1968, 6, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M.; Greer, J. Automatic identification of secondary structure in globular proteins. J. Mol. Biol. 1977, 114, 181–239. [Google Scholar] [CrossRef]
- Schwenke, K.D. Structural studies on native and chemically modified storage proteins from rapeseed (Brassica napus L.) and related plant proteins. Nahrung 1990, 34, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Krzyzaniak, A.; Burova, T.; Haertlé, T.; Barciszewski, J. The structure and properties of napin-seed storage protein from rape (Brassica napus L.). Food/Nahrung 1998, 42, 201–204. [Google Scholar] [CrossRef]
- Barciszewski, J.; Szymanski, M.; Haertle, T. Minireview: Analysis of rape seed napin structure and potential roles of the storage protein. J. Protein Chem. 2000, 19, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Pantoja-Uceda, D.; Bruix, M.; Gimenez-Gallego, G.; Rico, M.; Santoro, J. Solution structure of RicC3, a 2S albumin storage protein from Ricinus communis. Biochemistry 2003, 42, 13839–13847. [Google Scholar] [CrossRef] [PubMed]
- Folawiyo, Y.L.; Apenten, R.K.O. The effect of heat- and acid-treatment on the structure of rapeseed albumin (napin). Food Chem. 1997, 58, 237–243. [Google Scholar] [CrossRef]
- Mohamad Ramlan, B.M.S.; Maruyama, N.; Adachi, M.; Hontani, N.; Saka, S.; Kato, N.; Utsumi, S. Comparison of protein chemical and physicochemical properties of rapeseed cruciferin with those of soybean glycinin. J. Agric. Food Chem. 2002, 50, 7380–7385. [Google Scholar] [CrossRef]
- AOCS. AOCS official method Am 2-93, Determination of oil content in oilseeds. In Official Methods and Recommended Practices of the AOCS, 5th ed.; Firestone, D.E., Ed.; AOCS Press: Champaign, IL, USA, 1997. [Google Scholar]
- AOAC. AOAC Official Method 990.03, Protein (crude) in animal feed -combustion method. In Official Methods of Analysis of AOAC International, 18th ed.; Latimer, D.G.W., Jr., Horwitz, D.W., Eds.; AOAC International: Rockville, MD, USA, 2005. [Google Scholar]
- Laemmli, U.K. Glycine-SDS-PAGE for separation of proteins. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | pH | α-Helix | β-Sheet | β-Turn | Random | Surface Hydrophobicity |
|---|---|---|---|---|---|---|
| (%) | (%) | (%) | (%) | (S0) 2 | ||
| Cruciferin | 3 | 10.7 ±1.0 a | 25.4 ± 3.3 a | 26.0 ± 0.7 a | 38.0 ± 3.0 a | 6666.7 ± 47.2 a |
| 7 | 7.6 ± 0.7 b | 39.2 ± 1.9 b | 20.2 ± 0.9 b | 33.1 ± 1.6 b | 346.7 ± 6.4 b | |
| 10 | 4.8 ± 0.2 c | 18.4 ± 2.2 c | 26.3 ± 0.6 a | 50.5 ± 1.1 c | 208.0 ± 1.3 c | |
| Napin | 3 | 24.1 ± 0.7 a | NA | NA | 26.3 ± 1.3 a | 1239.3 ± 19.3 a |
| 7 | 27.5 ± 1.1 b | NA | NA | 26.9 ± 1.4 a | 103.6 ± 3.9 b | |
| 10 | 27.2 ± 0.7 a,b | NA | NA | 25.4 ± 0.6 a | 150.4 ± 1.6 c |
| pH | Denaturation Temperature, Tm (°C) | Enthalpy (J/g) | Onset of Peak (°C) | End of Peak (°C) |
|---|---|---|---|---|
| 3 | No peak was observed from 30 °C to 130 °C for both the species | |||
| 7 | 83.2 ± 0.8 a | 1.1 ± 0.3 a | 65–70 | 95–100 |
| 10 | 84.8 ± 0.2 a | 0.9 ± 0 a | 65–70 | 95–100 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, S.P.; McIntosh, T.C.; Wanasundara, J.P.D. Structural Properties of Cruciferin and Napin of Brassica napus (Canola) Show Distinct Responses to Changes in pH and Temperature. Plants 2016, 5, 36. https://doi.org/10.3390/plants5030036
Perera SP, McIntosh TC, Wanasundara JPD. Structural Properties of Cruciferin and Napin of Brassica napus (Canola) Show Distinct Responses to Changes in pH and Temperature. Plants. 2016; 5(3):36. https://doi.org/10.3390/plants5030036
Chicago/Turabian StylePerera, Suneru P., Tara C. McIntosh, and Janitha P. D. Wanasundara. 2016. "Structural Properties of Cruciferin and Napin of Brassica napus (Canola) Show Distinct Responses to Changes in pH and Temperature" Plants 5, no. 3: 36. https://doi.org/10.3390/plants5030036
APA StylePerera, S. P., McIntosh, T. C., & Wanasundara, J. P. D. (2016). Structural Properties of Cruciferin and Napin of Brassica napus (Canola) Show Distinct Responses to Changes in pH and Temperature. Plants, 5(3), 36. https://doi.org/10.3390/plants5030036