

Identification of Indigenous Thai Phlegmariurus Genotypic Population by Integrating Morphological and Molecular Studies




Abstract
1. Introduction
2. Results
2.1. Morphology and Cluster Analysis of Phlegmariurus Within Southern Thailand
2.1.1. Morphology Study
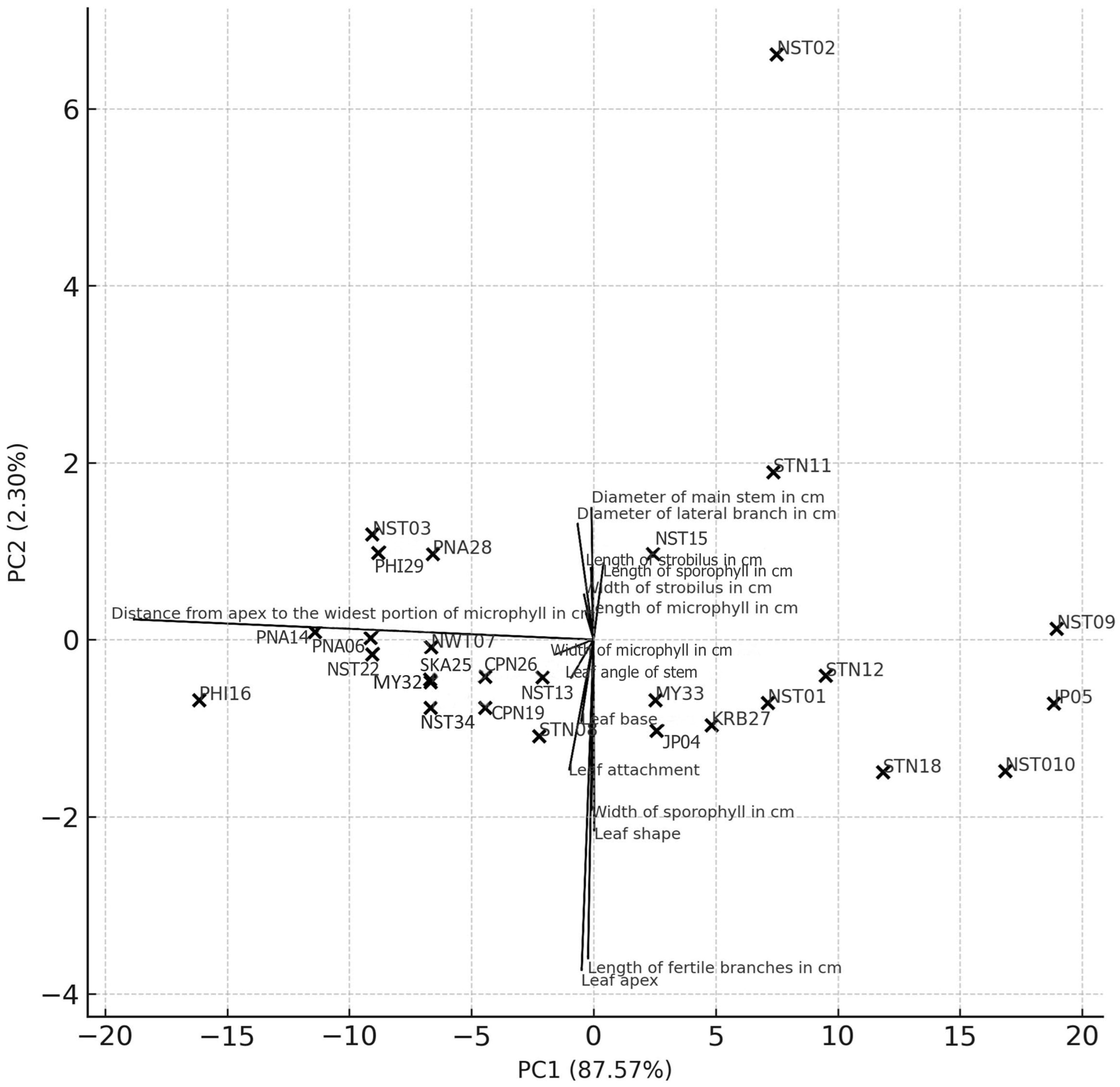
2.1.2. Principal Component Analysis (PCA) of Morphological Clustering in Phlegmariurus Specimens
2.1.3. PCA Biplot and Interpretation
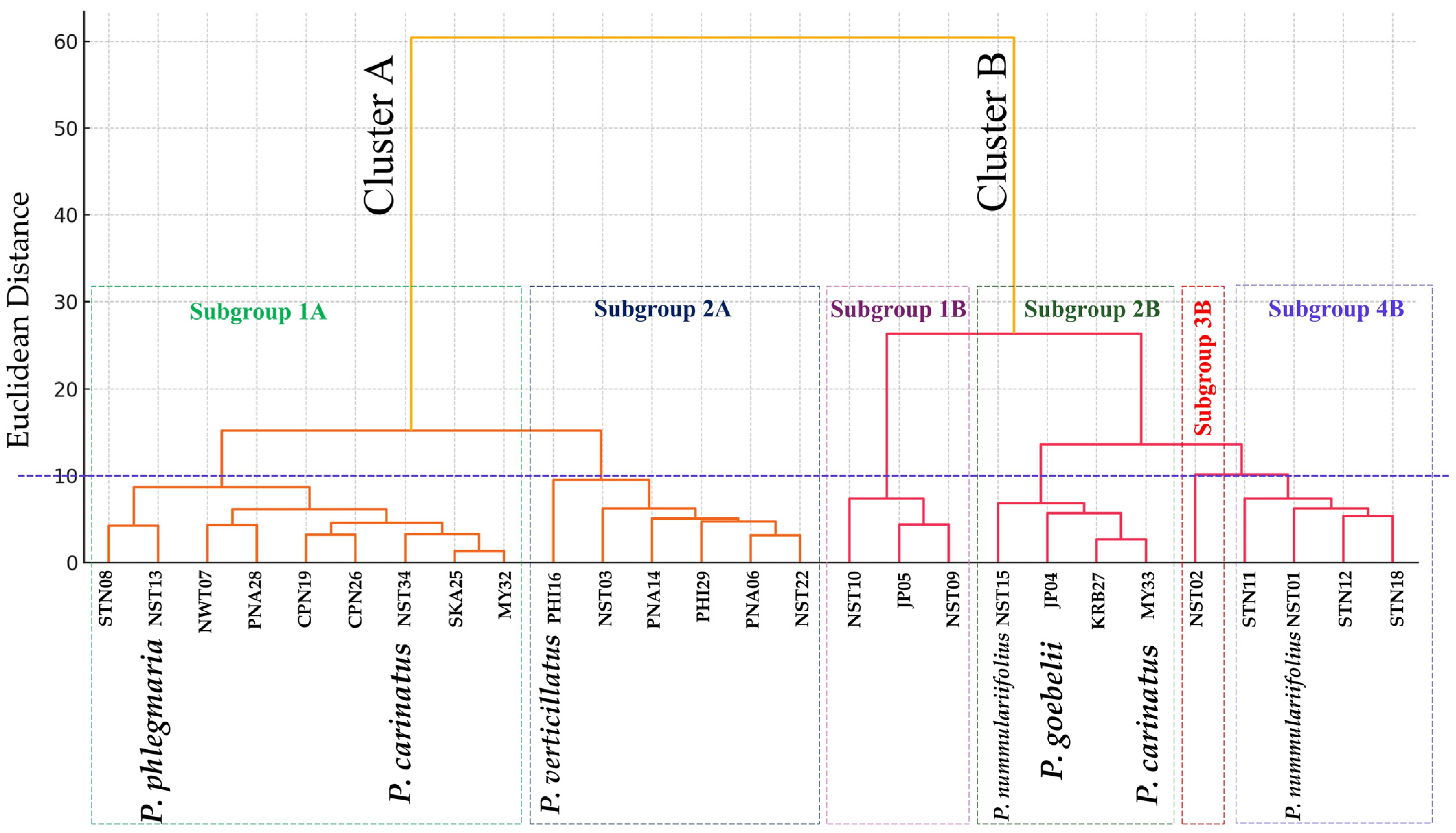
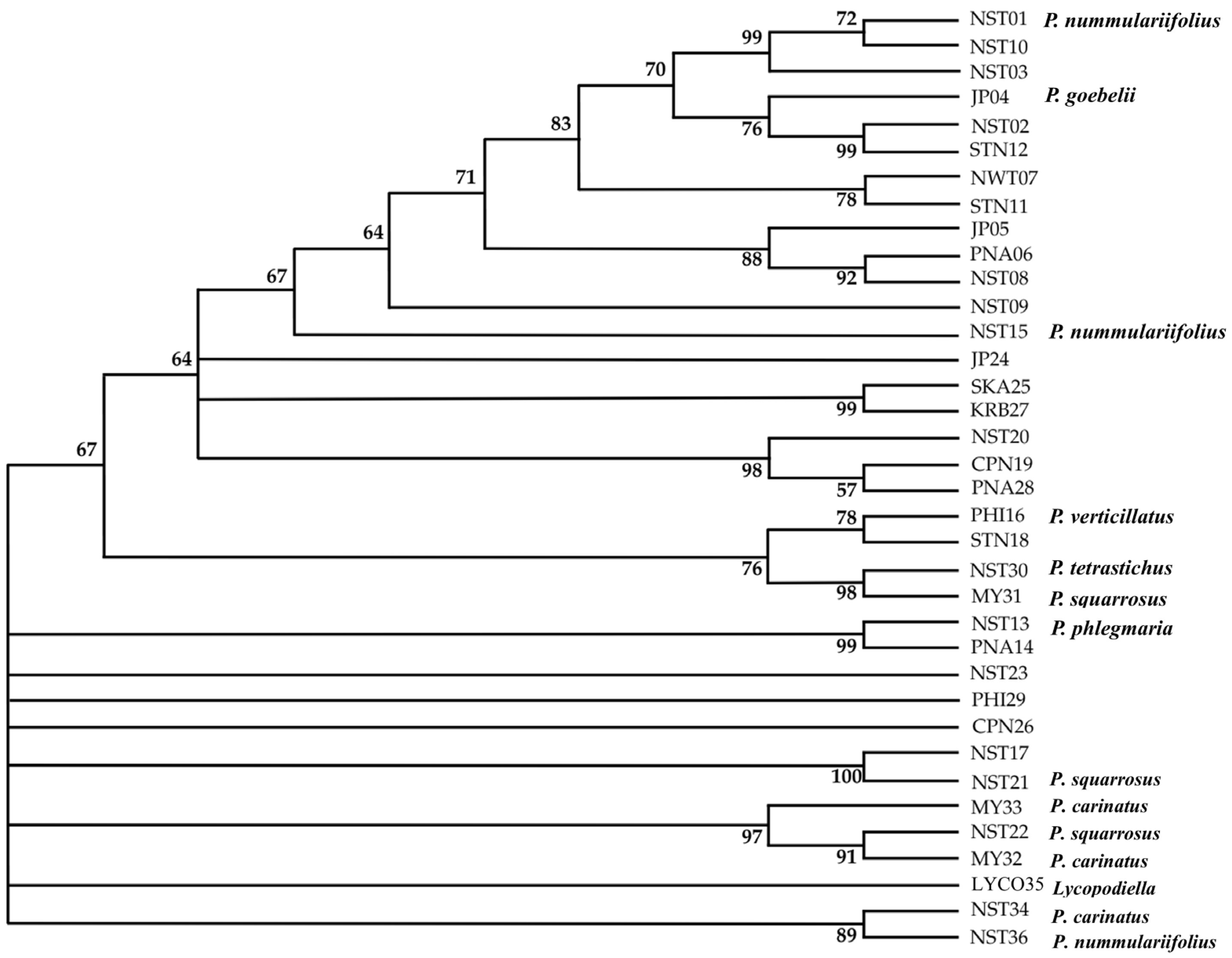
2.1.4. Hierarchical Clustering Analysis
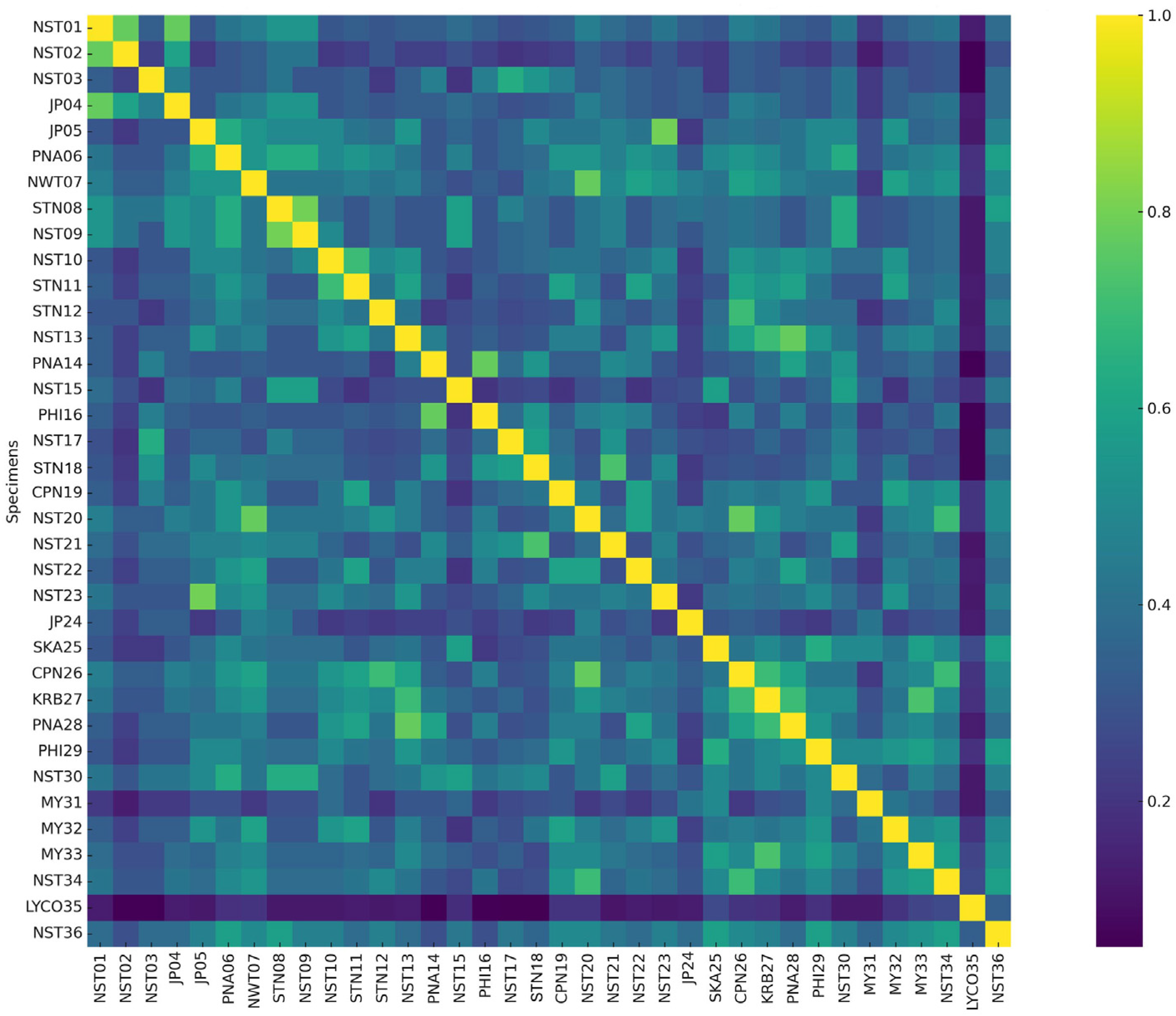
2.2. Analysis of Genetic Variability Among Phlegmariurus Species Using AFLP and SSR Marker
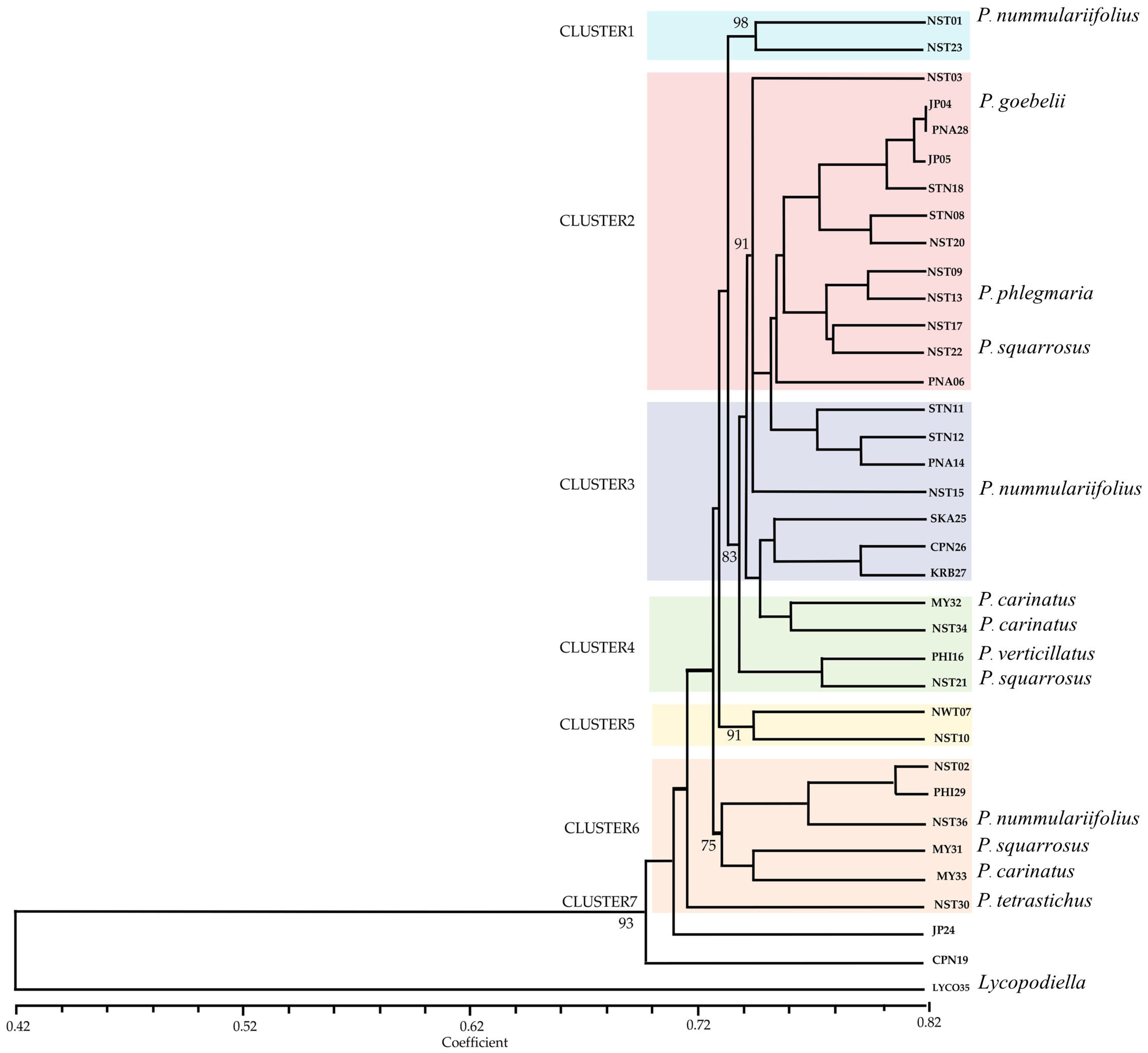
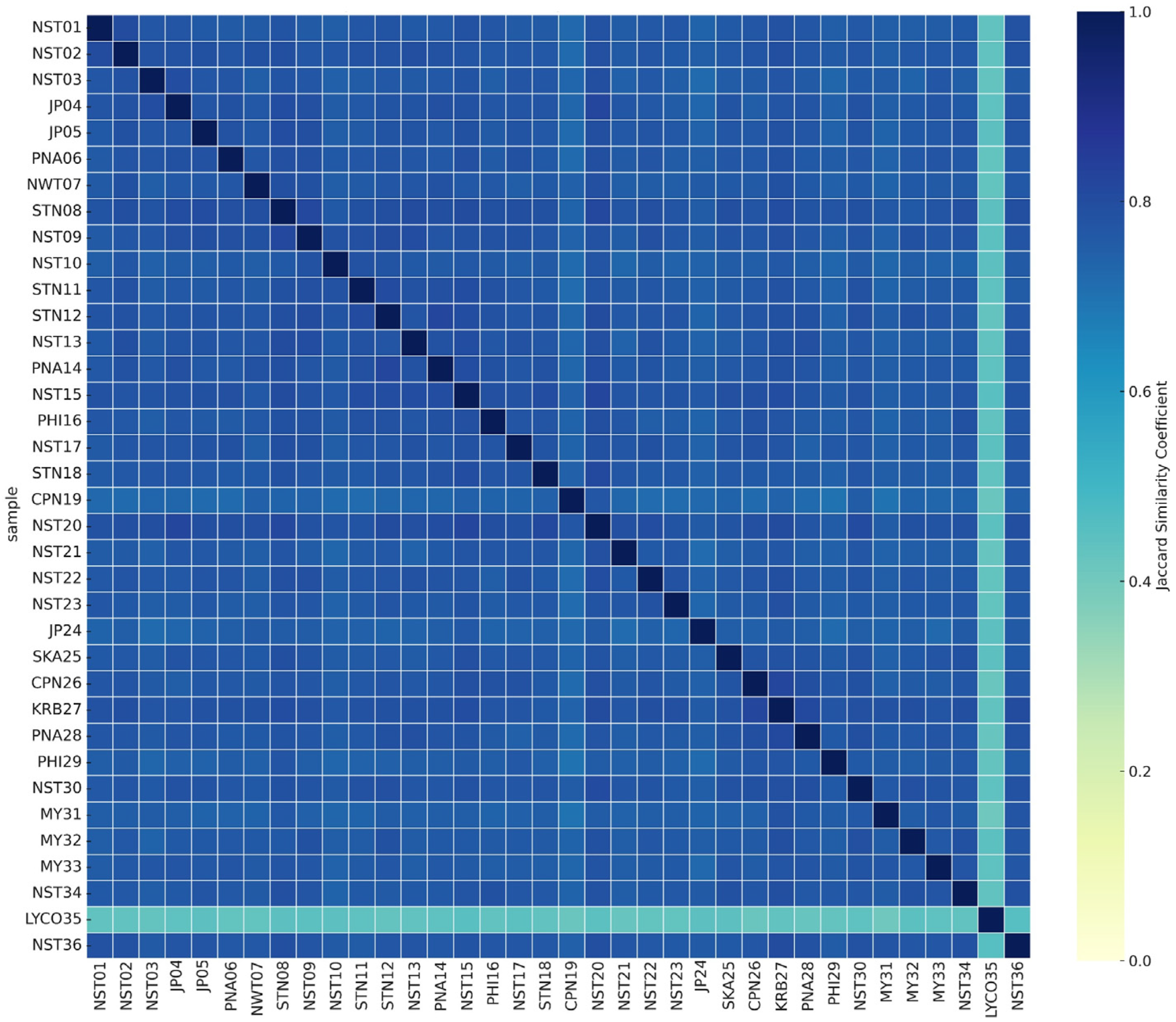
2.2.1. Amplified Fragment Length Polymorphism Analysis (AFLP)
2.2.2. Simple Sequence Repeat Analysis (SSR)
2.2.3. Efficiency of AFLP and SSR Markers for Phlegmariurus Diversity Assessment
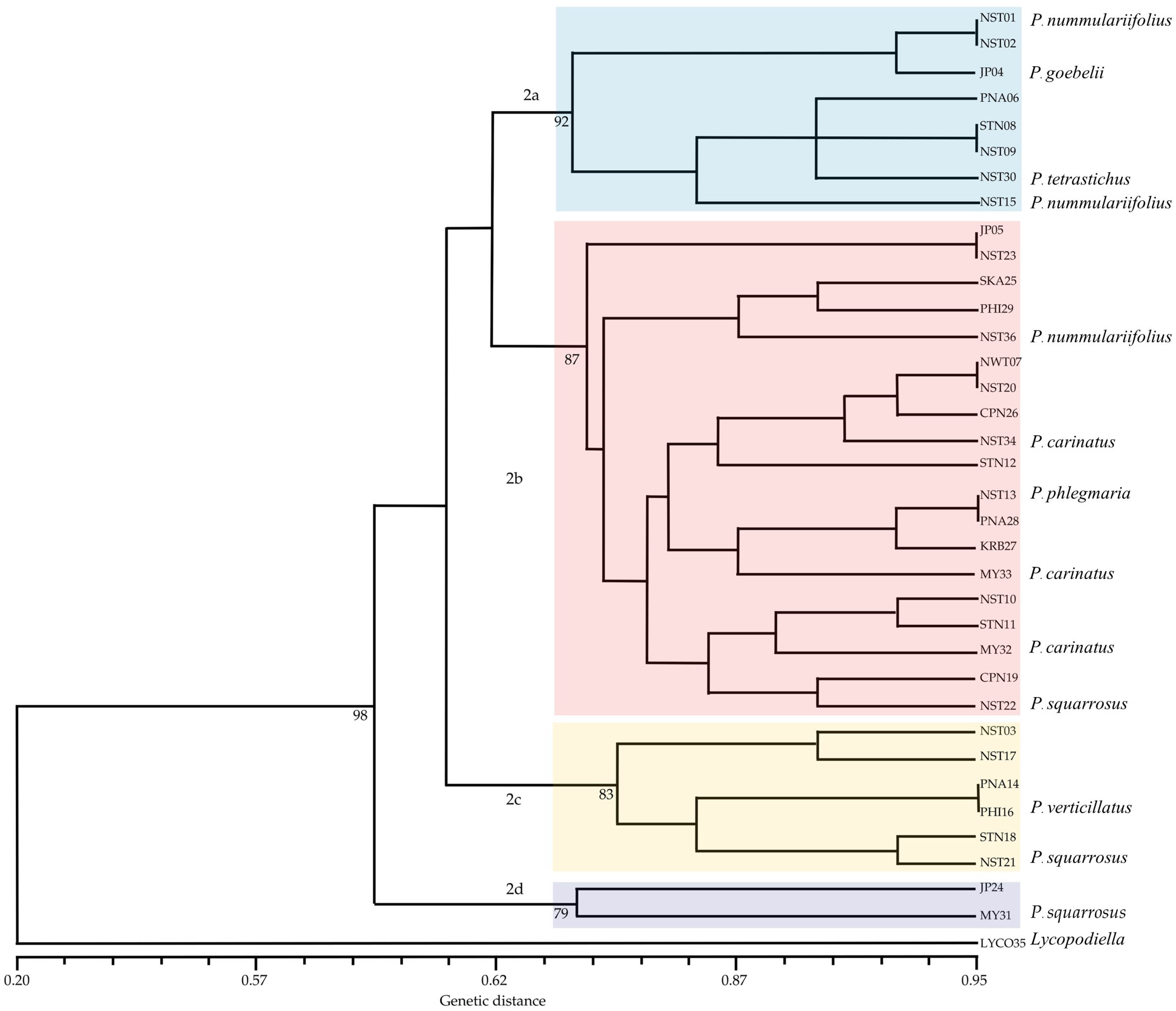
2.3. Molecular Phylogenetics of Phlegmariurus Based on Chloroplast rbcL and psbA-trnH Sequences
2.3.1. Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase Gene (rbcL)
2.3.2. psbA-trnH Intergenic Spacer
2.4. Descriptions of Three New Phlegmariurus Species, NST20, NST02, and STN11
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Morphology Study of Phlegmariurus
4.3. Analysis of Genetic Variability Among Phlegmariurus Species Using AFLP and SSR Marker
4.3.1. Total Genomic DNA Extraction
4.3.2. Genetic Variability Among Phlegmariurus Species Using AFLP Analysis
4.3.3. Genetic Variability Among Phlegmariurus Species Using SSR Analysis
4.3.4. Evaluating the Effectiveness of AFLP and SSR Markers for Genetic Diversity Analysis in Phlegmariurus
4.4. DNA Sequencing and Phylogenetic Analysis Using Chloroplast rbcL and psbA-trnH Sequences
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- PPG I. A community-derived classification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Boonkerd, T.; Klinratana, P.; Pollawath, R. Taxonomic revision of the Lycopodiaceae in Thailand. In Proceedings of the 6th Asian Fern Symposium, Bali, Indonesia, 26–28 August 2014. [Google Scholar] [CrossRef]
- Øllgaard, B. A revised classification of the Lycopodiaceae s. lat. Opera Bot. 1987, 92, 153–178. [Google Scholar]
- Tagawa, M.; Iwatsuki, K. Pteridophytes. In Flora Thailand; Smitiand, T., Larsen, K., Eds.; The Tistr Press: Bangkok, Thailand, 1979; Volume 3, p. 128. [Google Scholar]
- Parris, B.S.; Latiff, A. Towards a Pteridophyte Flora of Malaysia: A Provisional Checklist of Taxa. Malay. Nat. J. 1997, 50, 235–280. [Google Scholar]
- Holub, J. Transfers of Lycopodium species to Huperzia: With a note on generic classification in Huperziaceae. Folia Geobot. Phytotax. 1985, 20, 67–80. [Google Scholar] [CrossRef]
- Øllgaard, B. Index of the Lycopodiaceae. Biol. Skr. 1989, 34, 1–135. [Google Scholar]
- Øllgaard, B. Neotropical Lycopodiaceae—An overview. Ann. Mo. Bot. Gard. 1992, 79, 687–717. [Google Scholar] [CrossRef]
- Wikström, N.; Kenrick, P. Phylogeny of Lycopodiaceae (Lycopsida) and the relationships of Phylloglossum drummondii Kunze based on rbcL sequences. Int. J. Plant Sci. 1997, 158, 862–871. [Google Scholar] [CrossRef]
- Field, A.R.; Testo, W.; Bostock, P.D.; Holtum, J.A.M.; Waycott, M. Molecular phylogenetics and the morphology of the Lycopodiaceae subfamily Huperzioideae supports three genera: Huperzia, Phlegmariurus and Phylloglossum. Mol. Phylogenet. Evol. 2016, 94, 635–657. [Google Scholar] [CrossRef]
- Ching, R.C. The taxonomy of Chinese Lycopodiaceae (Sen. Lat.) I. Acta Bot. Yunnan 1981, 3, 1–9+291–305. [Google Scholar]
- Ching, R.C. The taxonomy of Chinese Lycopodiaceae (Sen. Lat.) III. Acta Bot. Yunnan 1982, 4, 119–128. [Google Scholar]
- Zhang, L.B.; Kung, H.S. A taxonomic study of Hupersia Bernh. (s.s.) sect. Hupersia in China. Acta Phytotax. Sin. 1998, 36, 521–529. [Google Scholar]
- Zhang, L.B.; Kung, H.S. Taxonomy of the genus Hupersia Bernh. (sen. str.) sect. Serratae (Rothm.) Holub in China. Acta Phytotax. Sin. 2000, 38, 13–22. [Google Scholar]
- Zhang, L.B.; Kung, H.S. On the taxonomy of Phlegmariurus (Herter) Holub sect. Huperzioides H.S. Kung et L. B. Zhang (sect. nov.) with notes on the infrageneric classification of the genus Phlegmariurus in China. Acta Phytotax. Sin. 1999, 37, 40–53. [Google Scholar]
- Zhang, L.B.; Kung, H.S. The reclassification of Lycopodiaceae (s. str.) in China. Acta Phytotax. Sin. B 2000, 38, 266–275. [Google Scholar]
- Wagner, W.H.; Beitel, J.M. Lycopodiaceae Mirbel, Club-moss Family. In Flora of North America; Flora of North America Association; Oxford University Press: New York, NY, USA, 1993; Volume 2, pp. 18–37. [Google Scholar]
- Hassler, M. World Ferns. Synonymic Checklist and Distribution of Ferns and Lycophytes of the World. (1994–2025): Version 25.02; Last Update February 13th. 2025. Available online: https://www.worldplants.de/world-ferns/ferns-and-lycophytes-list?name=Asplenium-dissectum&cHash=9ab4d4fae9d2626faf1d5644981bec45#plantUid-14042 (accessed on 14 February 2025).
- Yan, X.; Xiao, B.; Han, Y.; Yuan, W.; Shang, F. AFLP analysis of genetic relationships and diversity of some Chinese Osmanthus fragrans cultivars. Life Sci. J. 2008, 16, 11–16. [Google Scholar]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Van de Lee, T.; Hornes, M.; Frijters, A. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, U.; Yadav, H.K. Identification of EST-SSRs and molecular diversity analysis in Mentha piperita. Crop J. 2015, 3, 335–342. [Google Scholar] [CrossRef]
- Guo, B.; Ren, J.-Y.; He, M.-N.; Yao, K.; Wang, T.-S.; Wang, L.-Q.; Liu, X.; He, W.; Fu, Y.-P.; Wang, D.-L.; et al. Development of polymorphic simple sequence repeat markers in Huperzia serrata (Lycopodiaceae). Appl. Plant Sci. 2019, 7, e11273. [Google Scholar] [CrossRef]
- Letsiou, S.; Madesis, P.; Vasdekis, E.; Montemurro, C.; Grigoriou, M.E.; Skavdis, G.; Moussis, V.; Koutelidakis, A.E.; Tzakos, A.G. DNA barcoding as a plant identification method. Appl. Sci. 2024, 14, 1415. [Google Scholar] [CrossRef]
- Yang, J.; Vázquez, L.; Chen, X.; Li, H.; Zhang, H.; Liu, Z.; Zhao, G. Development of chloroplast and nuclear DNA markers for Chinese oaks (Quercus subgenus Quercus) and assessment of their utility as DNA barcodes. Front. Plant Sci. 2017, 8, 816. [Google Scholar] [CrossRef]
- Manhart, J.R. Phylogenetic analysis of green plant rbcL sequences. Mol. Phylogenet. Evol. 1994, 3, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.G.; Huo, K.K.; Wang, J.; Pan, S.L. A molecular phylogenetic study of Huperziaceae based on chloroplast rbcL and psbA-trnH sequences. J. Syst. Evol. 2008, 46, 213–219. [Google Scholar] [CrossRef]
- Yatsentyuk, S.P.; Valiejo-Roman, K.M.; Samigullin, T.H.; Wikström, N.; Troitsky, A.V. Evolution of Lycopodiaceae inferred from spacer sequencing of chloroplast rRNA genes. Russ. J. Genet. 2001, 37, 1274–1280. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA12: Molecular Evolutionary Genetics Analysis Version 12. Mol. Biol. Evol. 2024, 41, 3022–3027. [Google Scholar] [CrossRef]
- Liu, Y.C.; Su, J.; Wu, X.D.; Zhang, Z.J.; Fan, M.; Zhu, Q.F.; He, J.; Li, X.N.; Peng, L.Y.; Cheng, X.; et al. Five new Lycopodium alkaloids from the aerial parts of Phlegmariurus henryi. Fitoterapia 2016, 115, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P. Recently isolated lycodine-type Lycopodium alkaloids and their total systhesis: A review. Future J. Pharm. Sci. 2020, 6, 99. [Google Scholar] [CrossRef]
- Testo, A.L.; Sundue, M.A. A4 000 tip plastid phylogeny of the lycophytes reveals paleotropical origins and multiple intercontinental dispersals. Syst. Biol. 2016, 65, 200–211. [Google Scholar] [CrossRef]
- Kessler, M.; Smith, A.R. Taxonomic revision of Phlegmariurus (Lycopodiaceae) in Bolivia: Metric versus ordinal characters in species delimitation. Contrib. U. S. Natl. Herb. 2002, 45, 47–71. [Google Scholar]
- Jiang, R.-H.; Zhang, X.-C. Two new species of the lycophyte genus Phlegmariurus (Lycopodiaceae) from China. Turczaninowia 2022, 25, 131–142. [Google Scholar] [CrossRef]
- Kiew, R.; Kamin, I. Two new Phlegmariurus species (Lycopodiaceae) and one new combination from Peninsular Malaysia. PhytoKeys 2018, 96, 99–110. [Google Scholar] [CrossRef]
- Liang-Liang, Y.U.E.; Hang, S.U.N. Montane refugia isolation and plateau population expansion: Phylogeography of Sino-Himalayan endemic Spenceria ramalana (Rosaceae). J. Syst. Evol. 2014, 52, 326–340. [Google Scholar] [CrossRef]
- Krishnamurthy, S.L.; Prashanth, Y.; Rao, A.M.; Reddy, K.M. Assessment of AFLP marker based genetic diversity in chilli (Capsicum annuum L. & C. baccatum L.). Indian J. Biotechnol. 2015, 14, 49–54. [Google Scholar]
- Zhao, R.; He, Q.; Chu, X.; He, A.; Zhang, Y.; Zhu, Z. Regional environmental differences significantly affect the genetic diversity and differentiation of Carpinus tientaiensis. Front. Plant Sci. 2024, 15, 1277173. [Google Scholar] [CrossRef]
- de Carvalho, J.A.; Hagen, F.; Fisher, M.C.; de Camargo, Z.P.; Rodrigues, A.M. Genome-wide mapping using new AFLP markers to explore intraspecific variability in Sporothrix species. Front. Microbiol. 2020, 11, 7329091. [Google Scholar] [CrossRef]
- Brown-Guedira, G.L.; Thompson, J.A.; Nelson, R.L.; Warburton, M.L. Evaluation of genetic diversity of soybean introductions and North American ancestors using RAPD and SSR markers. Crop Sci. 2000, 40, 815–823. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, H.; Wu, X.; Ma, Y.; Zhang, J.; Li, Y.; Shao, G.; Chen, H.; Han, R.; Xu, Z. SSR marker-based analysis for identification and of genetic diversity of non-heading Chinese cabbage varieties. Front. Plant Sci. 2023, 14, 1112748. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.J.; Su, M.X.; Gao, H.; Yan, G.Y.; Li, S.S.; Chen, J.M.; Bai, Y.Y.; Deng, J.G. SSR marker-based genetic diversity and structure analyses of Camellia nitidissima var. phaeopubisperma from different populations. PeerJ 2025, 21, e18845. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Itoo, H.; Shah, R.A.; Qurat, S.; Khursheed, S.; Bhat, Z.A.; Mir, M.A.; Rather, G.H.; Zargar, S.M.; Shah, M.D.; Padder, B.A. Genome-wide characterization and development of SSR markers for genetic diversity analysis in northwestern Himalayas Walnut (Juglans regia L.). 3 Biotech 2023, 13, 136. [Google Scholar] [CrossRef]
- Misiukevičius, E.; Frercks, B.; Šikšnianienė, J.B.; Kącki, Z.; Gębala, M.; Akulytė, P.; Trilikauskaitė, E.; Stanys, V. Assessing the genetic diversity of daylily germplasm using SSR markers: Implications for daylily breeding. Plants 2023, 12, 1752. [Google Scholar] [CrossRef]
- Maras, M.; Sustar-Vozlic, J.; Javornik, B.; Meglic, V. The efficiency of the AFLP and SSR markers in genetic diversity estimation and gene pool classification of common bean (Phaseolus vulgaris L.). Acta Agric. Slov. 2008, 91, 87–96. [Google Scholar] [CrossRef]
- Étienne, L.-B.; Chen, B.-H.; Marie-Ève, G.-L.; Bruce, A.F.; Julian, R.S. RAD sequencing resolves the phylogeny, taxonomy and biogeography of Trichophoreae despite a recent rapid radiation (Cyperaceae). Mol. Phylogenet. Evol. 2020, 145, 106727. [Google Scholar] [CrossRef]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, A.J.; Burgess, K.S.; Kesanakurti, P.R.; Graham, S.W.; Newmaster, S.G.; Husband, B.C.; Hajibabaei, M.; Barrett, S.C.H. Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS ONE 2008, 3, e2802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Liu, C.; Shi, L.; Liu, R.; Liang, D.; Li, H.; Cherny, S.S.; Chen, S. Utility of the trnH-psbA intergenic spacer region and its combinations as plant DNA barcodes: A meta-analysis. PLoS ONE 2012, 7, e48833. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, X.Y.; Xie, C.; Liu, C.; Song, J.Y.; Yao, H.; Luo, K.; Zhu, Y.; Gao, T.; Pang, X.; Qian, J.; et al. Species identification of medicinal Pteridophytes by a DNA barcode marker, the chloroplast psbA-trnH intergenic region. Biol. Pharm. Bull. 2010, 33, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and Using a Plant DNA Barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef]
- Chen, D.K.; Zhou, X.M.; Rothfels, C.J.; Shepherd, L.D.; Knapp, R.; Zhang, L.; Lu, N.T.; Fan, X.P.; Wan, X.; Gao, X.F.; et al. A global phylogeny of Lycopodiaceae (Lycopodiales; lycophytes) with the description of a new genus, Brownseya, from Oceania. Taxon 2021, 71, 12597. [Google Scholar] [CrossRef]
- Nurmansyah; Alghamdi, S.S.; Migdadi, H.M.; Khan, M.A.; Afzal, M. AFLP-based analysis of variation and population structure in mutagenesis induced Faba Bean. Diversity 2020, 12, 303. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L. A Two-Locus Global DNA Barcode for Land Plants: The Coding rbcL Gene Complements the Non-Coding trnH-psbA Spacer Region. PLoS ONE 2007, 2, e508. [Google Scholar] [CrossRef] [PubMed]
- JASP Team. JASP, Version 0.19.3.0; Computer software; JASP Team: Amsterdam, The Netherlands, 2024.
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Masoudi, N.A.; Tonomura, K.; Kawashima, S.; Moriya, Y.; Suzuki, M.; Itoh, M.; Kanehisa, M.; Endo, T.; Goto, S.E. Gassembler: Online bioinformatics service for large-scale processing, clustering and assembling ESTs and genomic DNA fragments. Nucleic Acids Res. 2006, 34, W459–W462. [Google Scholar] [CrossRef]
- Jaccard, P. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 1908, 44, 223–270. [Google Scholar] [CrossRef]
- Rohlf, F.J. Numerical Taxonomy and Multivariate Analysis System, Version 2.11 Manual; Applied Biostatistics, Inc.: New York, NY, USA, 2000. [Google Scholar]
- Wikström, N.; Kenrick, P. Evolution of Lycopodiaceae (Lycopsida): Estimating divergence times from rbcL gene sequences by use of nonparametric rate smoothing. Mol. Phylogenet. Evol. 2001, 19, 177–186. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Bremer, K. The limits of amino acid sequence data in angiosperm phylogenetic reconstruction. Evolution 1988, 42, 795–803. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characters | Phlegmariurus | Lycopodiella | ||
|---|---|---|---|---|
| Mean (cm) | ±SD | Mean (cm) | ±SD | |
| Diameter of main stem | 4.17 | 2.40 | 0.60 | 0.00 |
| Diameter of lateral branch | 0.65 | 0.71 | 0.43 | 0.00 |
| Width of microphyll | 0.40 | 0.22 | 0.30 | 0.00 |
| Length of microphyll | 1.00 | 0.55 | 0.42 | 0.00 |
| Distance from apex to the widest portion of microphyll (mm) | 0.44 | 0.22 | 0.30 | 0.00 |
| Length of fertile branches | 12.60 | 14.92 | 1.20 | 0.00 |
| Length of strobilus | 8.41 | 4.11 | 0.21 | 0.00 |
| Width of strobilus | 0.33 | 0.27 | 0.08 | 0.00 |
| Length of sporophyll (mm) | 0.43 | 0.31 | 1.37 | 0.00 |
| Width of sporophyll (mm) | 0.29 | 0.31 | 0.13 | 0.00 |
| Trait | PC1 Loading | PC2 Loading |
|---|---|---|
| Distance from apex to the widest portion of microphyll (mm) | −0.99231 | 0.034427 |
| Width of microphyll (cm) | −0.08033 | −0.02468 |
| Leaf attachment (ordinal scale) | −0.0522 | −0.22074 |
| Leaf angle of stem (ordinal scale) | −0.04771 | −0.06406 |
| Diameter of lateral branch (cm) | −0.03444 | 0.196136 |
| Leaf base (ordinal scale) | −0.02633 | −0.13795 |
| Leaf apex (ordinal scale) | −0.02567 | −0.56209 |
| Width of strobilus (cm) | −0.02043 | 0.075919 |
| Length of microphyll (cm) | −0.01587 | 0.043359 |
| Length of fertile branches (cm) | −0.01199 | −0.54215 |
| Length of strobilus (cm) | −0.00559 | 0.121033 |
| Width of sporophyll (mm) | −0.00464 | −0.28928 |
| Diameter of main stem (cm) | −0.00462 | 0.223104 |
| Leaf shape (ordinal scale) | 0.001072 | −0.32471 |
| Primer Combination | Number of Total Loci Detected | Number of Polymorphic Loci | % of Polymorphic Loci | |
|---|---|---|---|---|
| E-AA | M-CAG | 92 | 92 | 100 |
| E-AG | M-CGA | 133 | 129 | 96.9 |
| E-AG | M-CGT | 85 | 73 | 85.9 |
| E-AT | M-CCA | 88 | 83 | 94.3 |
| E-AT | M-CTA | 96 | 91 | 94.8 |
| E-AT | M-CGA | 82 | 69 | 84.1 |
| E-ACC | M-CGT | 107 | 104 | 97.2 |
| E-AGA | M-CCA | 108 | 105 | 97.2 |
| E-AGA | M-CGT | 71 | 71 | 100 |
| E-AGC | M-CGT | 112 | 109 | 97.3 |
| Total | 974 | 926 | - | |
| Index | Marker System | |
|---|---|---|
| AFLP | SSR | |
| Number of assay units | 10.00 | 8.00 |
| Number of polymorphic bands | 926.00 | 44.00 |
| Number of monomorphic bands | 48.00 | 0 |
| Average number of polymorphic bands per assay unit | 92.60 | 5.50 |
| Number of loci | 974.00 | 44.00 |
| Number of loci per assay unit | 97.40 | 5.50 |
| Expected heterozygosity of the polymorphic loci | 0.33 | 0.35 |
| Fraction of polymorphic loci | 0.95 | 1.00 |
| Effective multiplex ratio | 92.60 | 5.50 |
| Marker index | 28.00 | 1.58 |
| Average PIC Value | 0.32 | 0.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chedao, N.; Pandey, A.C.; Suraninpong, P. Identification of Indigenous Thai Phlegmariurus Genotypic Population by Integrating Morphological and Molecular Studies. Plants 2025, 14, 1400. https://doi.org/10.3390/plants14091400
Chedao N, Pandey AC, Suraninpong P. Identification of Indigenous Thai Phlegmariurus Genotypic Population by Integrating Morphological and Molecular Studies. Plants. 2025; 14(9):1400. https://doi.org/10.3390/plants14091400
Chicago/Turabian StyleChedao, Nusanisa, Avinash Chandra Pandey, and Potjamarn Suraninpong. 2025. "Identification of Indigenous Thai Phlegmariurus Genotypic Population by Integrating Morphological and Molecular Studies" Plants 14, no. 9: 1400. https://doi.org/10.3390/plants14091400
APA StyleChedao, N., Pandey, A. C., & Suraninpong, P. (2025). Identification of Indigenous Thai Phlegmariurus Genotypic Population by Integrating Morphological and Molecular Studies. Plants, 14(9), 1400. https://doi.org/10.3390/plants14091400