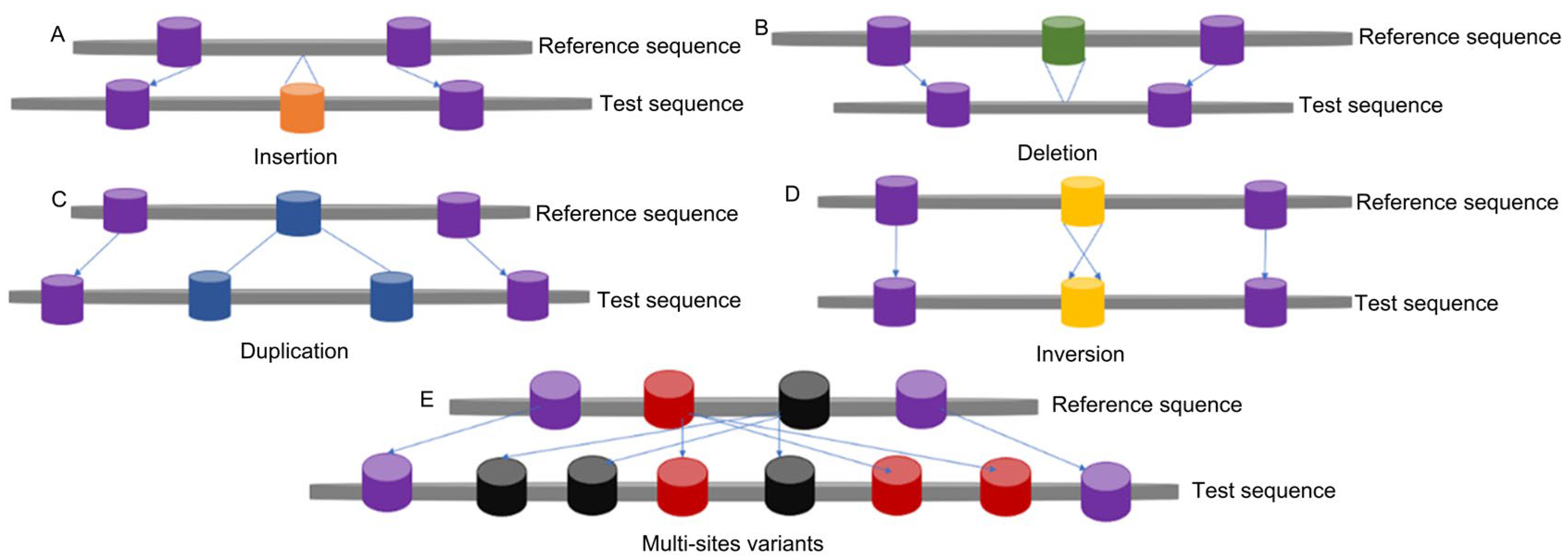
A Systematic Review of the Advances and New Insights into Copy Number Variations in Plant Genomes

 ,
,
Abstract
1. Introduction
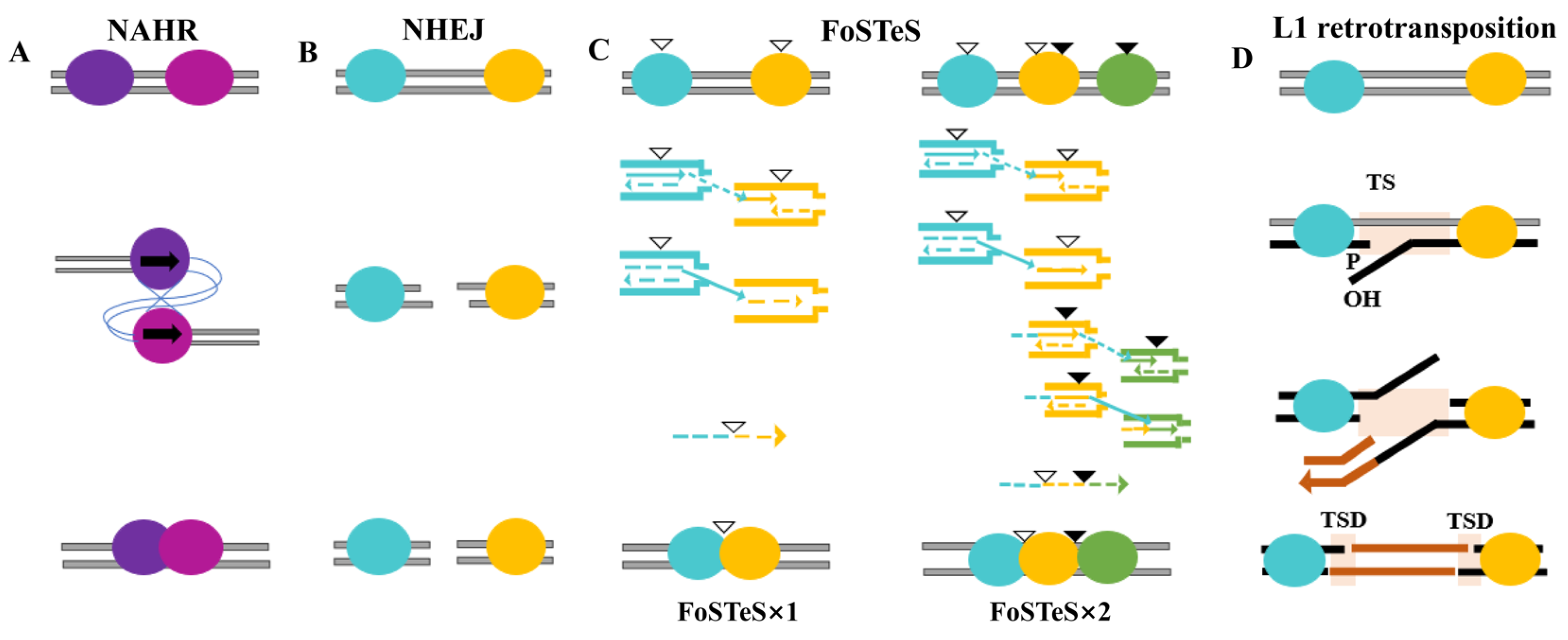
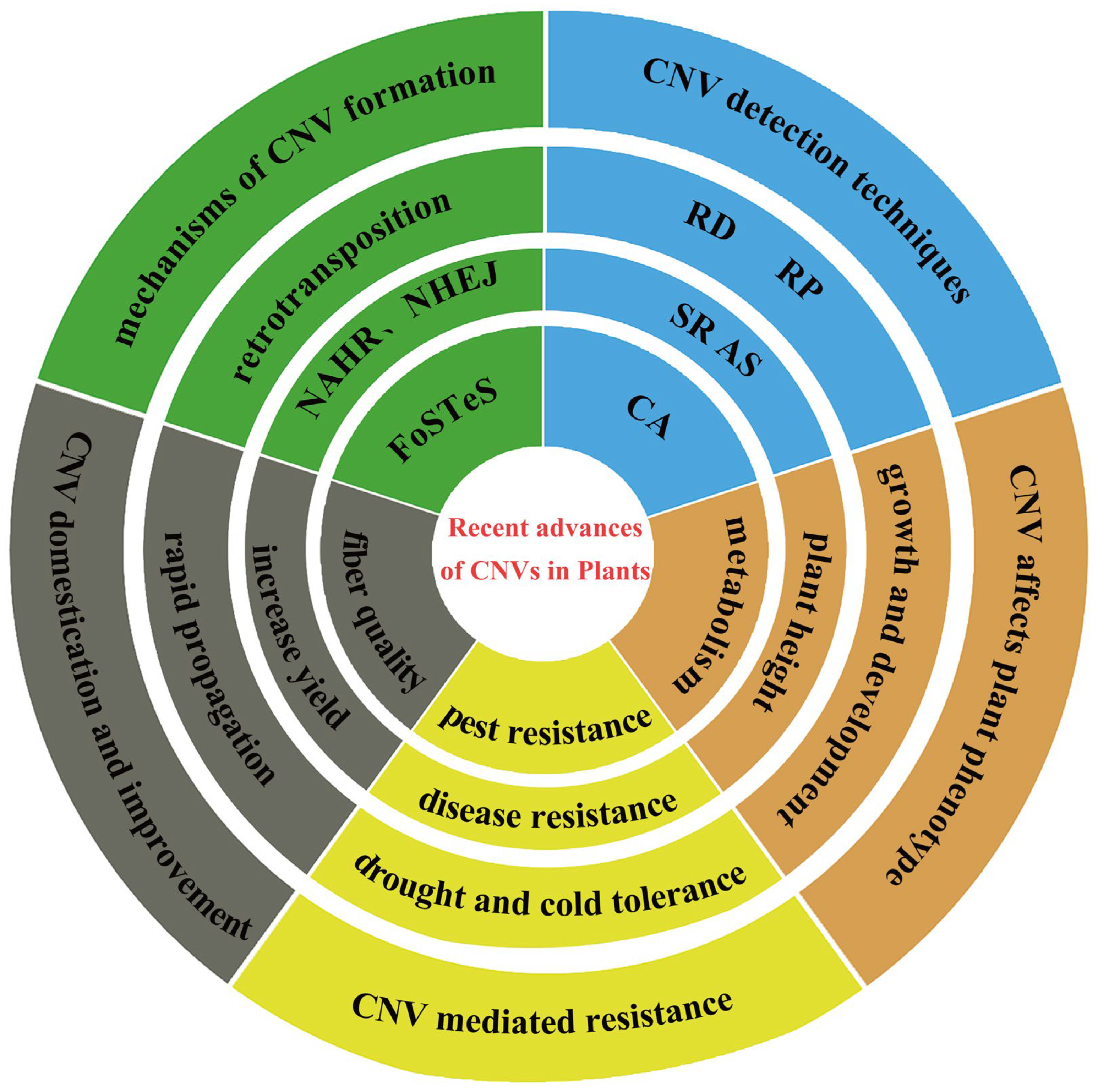
2. Mechanisms of CNV Formation
2.1. Non-Allelic Homologous Recombination (NAHR)
2.2. Non-Homologous End Joining (NHEJ)
2.3. The Fork Stalling and Template Switching (FoSTeS)
2.4. L1 Retrotransposition
3. Methods for Detecting CNVs
3.1. The Development History of Sequencing Technologies
3.2. CNV Identification Algorithms and Tools

{kind=link}
{kind=link}
{kind=link}
| Software | Language | Name | Year | References |
|---|---|---|---|---|
| PEMer (-) | Python/Perl | RP | 2009 | [64] |
| BreakDancer (Version 1.4.5) | Perl/C++ | RP | 2009 | [65] |
| SegSeq (-) | MatLab | RD | 2009 | [88] |
| Pindel (Version 0.2.5b9) | C++ | SR | 2009 | [68] |
| mrFAST (Version 2.6.1) | C | RD | 2009 | [89] |
| CNV-seq (Version 0.9.7) | Perl/R | RD | 2009 | [90] |
| RDXplorer (Version 3.2) | Python/R | RD | 2009 | [91] |
| SV Detect (Version 1.4) | Perl | SR | 2010 | [71] |
| rSW-seq (-) | C | RD | 2010 | [92] |
| cnD (-) | D | SR | 2010 | [93] |
| CNVer (-) | C++ | CA | 2010 | [69] |
| GenomeSTRiP (Version 2.0) | Java/R | CA | 2011 | [70] |
| BIC-Seq (Version 0.2.4) | Perl/R | RD | 2011 | [60] |
| CNVnator (Version 0.4.1) | C++; Perl | RD | 2011 | [61] |
| ReadDepth (Version 0.9.8.1) | R | RD | 2011 | [62] |
| JointSLM (-) | R | RD | 2011 | [83] |
| PRISM (Version 1.1.6) | C | SR | 2012 | [66] |
| SVseq2 (Version 2.2) | C++ | SR | 2012 | [67] |
| ERDS (Version 1.1) | C | RD | 2012 | [94] |
| DELLY (Version 1.1.3) | C++/R | CA | 2012 | [72] |
| GASVPro (Version SCR_005259) | C++ | CA | 2012 | [95] |
| Control-FREEC (Version 11.6) | C++ | RD | 2012 | [79] |
| CnvHiTSeq (Version 0.1.2) | Java | CA | 2012 | [87] |
| Cn.MOPS (Version 1.38.0) | R | RD | 2012 | [63] |
| Magnolya (Version 0.15) | Python | AS | 2012 | [59] |
| Clever-sv (-) | C++ | CA | 2013 | [96] |
| SoftSearch (Version SCR_006683) | Perl | CA | 2013 | [97] |
| CNVeM (Version 0.710) | C | RD | 2013 | [80] |
| CNVrd2 (Version 3.21) | R | RD | 2014 | [98] |
| Gindel (Version 0.8) | C++ | CA | 2014 | [99] |
| PSCC (-) | Perl | CA | 2014 | [100] |
| LUMPY (Version 0.3.1) | C; C++; Python; Shell | CA | 2014 | [101] |
| Hydra-Multi (Version 0.5.4) | C++ | CA | 2015 | [102] |
| CNVcaller (Version 1.0) | Python | CA | 2017 | [103] |
| GATK4 (Version 4.3.0.0) | Java | CA | 2018 | [104] |
| Hecaton (-) | Python | RD | 2019 | [74] |
| ExomeDepth (Version 1.1.16) | R | RD | 2020 | [81] |
| CONY (-) | R | CA | 2020 | [105] |
| inCNV (Version 2.2.0) | Python | RD | 2020 | [106] |
| CNVpytor (Version 1.2.2) | Python | CA | 2021 | [77] |
| Svpluscnv (-) | R | CA | 2021 | [82] |
| SCYN (-) | R | CA | 2021 | [84] |
| SCSilicon (-) | Python | CA | 2022 | [78] |
4. Recent Advances of CNVs in Plant Genomes
4.1. CNVs Affect the Phenotype of Plants
4.2. CNVs Enhance Plant Tolerance to Adverse Environments
4.3. CNVs Accelerate the Domestication of Plants
4.4. CNVs Promote Genetic Improvement in Plants
5. Future Prospects
5.1. Enhanced Detection Sensitivity and Accuracy
5.2. Integrating Multi-Omics Data to Study CNV
5.3. Artificial Intelligence Revolutionizes CNV Detection
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, H.J.; Zhang, D.Q. Copy number variations in plant genomes. Mol. Plant Breed. 2015, 13, 1895–1910. [Google Scholar] [CrossRef]
- Feuk, L.; Marshall, C.R.; Wintle, R.F.; Scherer, S.W. Structural variants: Changing the landscape of chromosomes and design of disease studies. Hum. Mol. Genet. 2006, 15, R57–R66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The database of genomic variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- Moradi, M.H.; Mahmodi, R.; Farahani, A.H.K.; Karimi, M.O. Genome-wide evaluation of copy gain and loss variations in three Afghan sheep breeds. Sci. Rep. 2022, 12, 14286. [Google Scholar] [CrossRef]
- Luo, X.; Cai, G.; Mclain, A.C.; Amos, C.I.; Cai, B.; Xiao, F. BMI-CNV: A Bayesian framework for multiple genotyping platforms detection of copy number variants. Genetics 2022, 222, iyac147. [Google Scholar] [CrossRef]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA copy number variation: Main characteristics, evolutionary significance, and pathological aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Mehta, D.; Iwamoto, K.; Ueda, J.; Bundo, M.; Adati, N.; Kojima, T.; Kato, T. Comprehensive survey of CNVs influencing gene expression in the human brain and its implications for pathophysiology. Neurosci. Res. 2014, 79, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, Y.; Liu, S.; Meng, Q. Genome-Wide Assessment Characteristics of Genes Overlapping Copy Number Variation Regions in Duroc Purebred Population. Front. Genet. 2021, 12, 753748. [Google Scholar] [CrossRef]
- Iafrate, A.J.; Feuk, L.; Rivera, M.N.; Listewnik, M.L.; Donahoe, P.K.; Qi, Y.; Scherer, S.W.; Lee, C. Detection of large-scale variation in the human genome. Nat. Genet. 2004, 36, 949–951. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Troge, J.; Alexander, J.; Young, J.; Lundin, P.; Månér, S.; Massa, H.; Walker, M.; Chi, M.; et al. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar] [CrossRef]
- Sudmant, P.H.; Mallick, S.; Nelson, B.J.; Hormozdiari, F.; Krumm, N.; Huddleston, J.; Coe, B.P.; Baker, C.; Nordenfelt, S.; Bamshad, M. Global diversity, population stratification, and selection of human copy-number variation. Science 2015, 349, aab3761. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kulkarni, H.; Bolivar, H.; Mangano, A.; Sanchez, R.; Catano, G.; Nibbs, R.J.; Freedman, B.I.; Quinones, M.P.; Bamshad, M.J.; et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005, 307, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Le Meur, N.; Laquerrière, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.A.; Shen, Y.; Korn, J.M.; Arking, D.E.; Miller, D.T.; Fossdal, R.; Saemundsen, E.; Stefansson, H.; Ferreira, M.A.; Green, T.; et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008, 358, 667–675. [Google Scholar] [CrossRef]
- Stefansson, H.; Rujescu, D.; Cichon, S.; Pietiläinen, O.P.; Ingason, A.; Steinberg, S.; Fossdal, R.; Sigurdsson, E.; Sigmundsson, T.; Buizer-Voskamp, J.E.; et al. Large recurrent microdeletions associated with schizophrenia. Nature 2008, 455, 232–236. [Google Scholar] [CrossRef]
- Bae, J.S.; Cheong, H.S.; Kim, L.H.; NamGung, S.; Park, T.J.; Chun, J.Y.; Kim, J.Y.; Pasaje, C.F.; Lee, J.S.; Shin, H.D. Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genom. 2010, 11, 232. [Google Scholar] [CrossRef]
- Kim, Y.M.; Ha, S.J.; Seong, H.S.; Choi, J.Y.; Baek, H.J.; Yang, B.C.; Choi, J.W.; Kim, N.Y. Identification of copy number variations in four horse breed populations in South Korea. Animals 2022, 12, 3501. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Tchinda, J.; McGrath, S.D.; Zhang, J.; Picker, S.R.; Cáceres, A.M.; Iafrate, A.J.; Tyler-Smith, C.; Scherer, S.W.; Eichler, E.E.; et al. Hotspots for copy number variation in chimpanzees and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 8006–8011. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, D.A.; van Heyningen, V. Long-range control of gene expression: Emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005, 76, 8–32. [Google Scholar] [CrossRef]
- Mitchell-Olds, T.; Schmitt, J. Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature 2006, 441, 947–952. [Google Scholar] [CrossRef]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; de Grassi, A.; Lee, C.; et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef]
- Cao, J.; Schneeberger, K.; Ossowski, S.; Günther, T.; Bender, S.; Fitz, J.; Koenig, D.; Lanz, C.; Stegle, O.; Lippert, C.; et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 2011, 43, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Springer, N.M.; Ying, K.; Fu, Y.; Ji, T.; Yeh, C.T.; Jia, Y.; Wu, W.; Richmond, T.; Kitzman, J.; Rosenbaum, H.; et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009, 5, e1000734. [Google Scholar] [CrossRef]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar] [CrossRef]
- Yu, P.; Wang, C.H.; Xu, Q.; Feng, Y.; Yuan, X.P.; Yu, H.Y.; Wang, Y.P.; Tang, S.X.; Wei, X.H. Genome-wide copy number variations in Oryza sativa L. BMC Genom. 2013, 14, 649. [Google Scholar] [CrossRef]
- Muñoz-Amatriaín, M.; Eichten, S.R.; Wicker, T.; Richmond, T.A.; Mascher, M.; Steuernagel, B.; Scholz, U.; Ariyadasa, R.; Spannagl, M.; Nussbaumer, T.; et al. Distribution, functional impact, and origin mechanisms of copy number variation in the barley genome. Genome Biol. 2013, 14, R58. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, L.; Chen, H.; Bu, F.; Li, G.; Sun, J.; Li, S.; Sun, H.; Jiao, C.; Blakely, R.; et al. Genome-Wide Mapping of Structural Variations Reveals a Copy Number Variant That Determines Reproductive Morphology in Cucumber. Plant Cell 2015, 27, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.G.; Diers, B.W.; Hudson, M.E. An efficient method for measuring copy number variation applied to improvement of nematode resistance in soybean. Plant J. 2016, 88, 143–153. [Google Scholar] [CrossRef]
- Hardigan, M.A.; Crisovan, E.; Hamilton, J.P.; Kim, J.; Laimbeer, P.; Leisner, C.P.; Manrique-Carpintero, N.C.; Newton, L.; Pham, G.M.; Vaillancourt, B.; et al. Genome Reduction Uncovers a Large Dispensable Genome and Adaptive Role for Copy Number Variation in Asexually Propagated Solanum tuberosum. Plant Cell 2016, 28, 388–405. [Google Scholar] [CrossRef]
- Zmienko, A.; Marszalek-Zenczak, M.; Wojciechowski, P.; Samelak-Czajka, A.; Luczak, M.; Kozlowski, P.; Karlowski, W.M.; Figlerowicz, M. AthCNV: A Map of DNA Copy Number Variations in the Arabidopsis Genome. Plant Cell 2020, 32, 1797–1819. [Google Scholar] [CrossRef]
- Zhao, Z.; Ding, Z.; Huang, J.; Meng, H.; Zhang, Z.; Gou, X.; Tang, H.; Xie, X.; Ping, J.; Xiao, F.; et al. Copy number variation of the restorer Rf4 underlies human selection of three-line hybrid rice breeding. Nat. Commun. 2023, 14, 7333. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.H.; Wright, S.J.; Small, L.L.; Olsen, K.M. De novo genome assembly of white clover (Trifolium repens L.) reveals the role of copy number variation in rapid environmental adaptation. BMC Biol. 2024, 22, 165. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Bieker, V.C.; Boheemen, L.V.; Connallon, T.; Martin, M.D.; Battlay, P.; Hodgins, K.A. Copy number variation contributes to parallel local adaptation in an invasive plant. Proc. Natl. Acad. Sci. USA 2025, 122, e2413587122. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002, 18, 74–82. [Google Scholar] [CrossRef]
- Bailey, J.A.; Eichler, E.E. Primate segmental duplications: Crucibles of evolution, diversity and disease. Nat. Rev. Genet. 2006, 7, 552–564. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Lupski, J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010, 61, 437–455. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Toffolatti, L.; Cardazzo, B.; Nobile, C.; Danieli, G.A.; Gualandi, F.; Muntoni, F.; Abbs, S.; Zanetti, P.; Angelini, C.; Ferlini, A.; et al. Investigating the mechanism of chromosomal deletion: Characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics 2002, 80, 523–530. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Zhang, F.; Liu, P.; Patel, A.; Sahoo, T.; Bacino, C.A.; Shaw, C.; Peacock, S.; Pursley, A.; Tavyev, Y.J.; et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009, 18, 2188–2203. [Google Scholar] [CrossRef] [PubMed]
- Cocquempot, O.; Brault, V.; Babinet, C.; Herault, Y. Fork stalling and template switching as a mechanism for polyalanine tract expansion affecting the DYC mutant of HOXD13, a new murine model of synpolydactyly. Genetics 2009, 183, 23–30. [Google Scholar] [CrossRef]
- Kidd, J.M.; Graves, T.; Newman, T.L.; Fulton, R.; Hayden, H.S.; Malig, M.; Kallicki, J.; Kaul, R.; Wilson, R.K.; Eichler, E.E. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell 2010, 143, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Ostertag, E.M.; Kazazian, H.H., Jr. Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 2001, 35, 501–538. [Google Scholar] [CrossRef]
- Goodier, J.L.; Kazazian, H.H., Jr. Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell 2008, 135, 23–35. [Google Scholar] [CrossRef]
- Verma, M.; Kulshrestha, S.; Puri, A. Genome sequencing. Methods Mol. Biol. 2017, 1525, 3–33. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Leamon, J.H. The development and impact of 454 sequencing. Nat. Biotechnol. 2008, 26, 1117–1124. [Google Scholar] [CrossRef]
- Green, R.E.; Briggs, A.W.; Krause, J.; Prüfer, K.; Burbano, H.A.; Siebauer, M.; Lachmann, M.; Pääbo, S. The neandertal genome and ancient DNA authenticity. Embo J. 2009, 28, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Akintunde, O.; Tucker, T.; Carabetta, V.J. The evolution of next-generation sequencing technologies. arXiv 2023, arXiv:2305.08724. [Google Scholar]
- Check Hayden, E. Genome sequencing: The third generation. Nature 2009, 457, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Rutkowska, L.; Pinkier, I.; Sałacińska, K.; Kępczyński, Ł.; Salachna, D.; Lewek, J.; Banach, M.; Matusik, P.; Starostecka, E.; Lewiński, A.; et al. Identification of New Copy number variation and the evaluation of a CNV detection tool for NGS panel data in polish familial hypercholesterolemia patients. Genes 2022, 13, 1424. [Google Scholar] [CrossRef]
- Wang, H.; Nettleton, D.; Ying, K. Copy number variation detection using next generation sequencing read counts. BMC Bioinform. 2014, 15, 109. [Google Scholar] [CrossRef]
- Nijkamp, J.F.; van den Broek, M.A.; Geertman, J.M.; Reinders, M.J.; Daran, J.M.; de Ridder, D. De novo detection of copy number variation by co-assembly. Bioinformatics 2012, 28, 3195–3202. [Google Scholar] [CrossRef]
- Xi, R.; Hadjipanayis, A.G.; Luquette, L.J.; Kim, T.M.; Lee, E.; Zhang, J.; Johnson, M.D.; Muzny, D.M.; Wheeler, D.A.; Gibbs, R.A.; et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc. Natl. Acad. Sci. USA 2011, 108, E1128–E1136. [Google Scholar] [CrossRef]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Hampton, O.; Coarfa, C.; Milosavljevic, A. ReadDepth: A parallel R package for detecting copy number alterations from short sequencing reads. PLoS ONE 2011, 6, e16327. [Google Scholar] [CrossRef] [PubMed]
- Klambauer, G.; Schwarzbauer, K.; Mayr, A.; Clevert, D.A.; Mitterecker, A.; Bodenhofer, U.; Hochreiter, S. cn.MOPS: Mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 2012, 40, e69. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Abyzov, A.; Mu, X.J.; Carriero, N.; Cayting, P.; Zhang, Z.; Snyder, M.; Gerstein, M.B. PEMer: A computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009, 10, R23. [Google Scholar] [CrossRef]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Y.; Brudno, M. PRISM: Pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants. Bioinformatics 2012, 28, 2576–2583. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Wu, Y. An improved approach for accurate and efficient calling of structural variations with low-coverage sequence data. BMC Bioinform. 2012, 13 (Suppl. S6), S6. [Google Scholar] [CrossRef]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Medvedev, P.; Fiume, M.; Dzamba, M.; Smith, T.; Brudno, M. Detecting copy number variation with mated short reads. Genome Res. 2010, 20, 1613–1622. [Google Scholar] [CrossRef]
- Handsaker, R.E.; Korn, J.M.; Nemesh, J.; McCarroll, S.A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011, 43, 269–276. [Google Scholar] [CrossRef]
- Zeitouni, B.; Boeva, V.; Janoueix-Lerosey, I.; Loeillet, S.; Legoix-né, P.; Nicolas, A.; Delattre, O.; Barillot, E. SVDetect: A tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics 2010, 26, 1895–1896. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]
- Hormozdiari, F.; Alkan, C.; Eichler, E.E.; Sahinalp, S.C. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009, 19, 1270–1278. [Google Scholar] [CrossRef]
- Wijfjes, R.Y.; Smit, S.; de Ridder, D. Hecaton: Reliably detecting copy number variation in plant genomes using short read sequencing data. BMC Genom. 2019, 20, 818. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Aguilar, S.; Vendrell, J.A.; Van Goethem, C.; Brousse, M.; Gozé, C.; Frantz, L.; Solassol, J. ifCNV: A novel isolation-forest-based package to detect copy-number variations from various targeted NGS datasets. Mol. Ther. Nucleic Acids 2022, 30, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, S.; Yuan, X. HBOS-CNV: A New Approach to Detect Copy Number Variations from Next-Generation Sequencing Data. Front. Genet. 2021, 12, 642473. [Google Scholar] [CrossRef]
- Suvakov, M.; Panda, A.; Diesh, C.; Holmes, I.; Abyzov, A. CNVpytor: A tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing. Gigascience 2021, 10, giab074. [Google Scholar] [CrossRef]
- Feng, X.; Chen, L. SCSilicon: A tool for synthetic single-cell DNA sequencing data generation. BMC Genom. 2022, 23, 359. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Wang, Z.; Hormozdiari, F.; Yang, W.Y.; Halperin, E.; Eskin, E. CNVeM: Copy number variation detection using uncertainty of read mapping. J. Comput. Biol. 2013, 20, 224–236. [Google Scholar] [CrossRef]
- Bretani, G.; Rossini, L.; Ferrandi, C.; Russell, J.; Waugh, R.; Kilian, B.; Bagnaresi, P.; Cattivelli, L.; Fricano, A. Segmental duplications are hot spots of copy number variants affecting barley gene content. Plant J. 2020, 103, 1073–1088. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Egolf, L.E.; Giorgi, F.M.; Diskin, S.J.; Margolin, A.A. svpluscnv: Analysis and visualization of complex structural variation data. Bioinformatics 2021, 37, 1912–1914. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Benelli, M.; Yoon, S.; Roviello, F.; Torricelli, F. Detecting common copy number variants in high-throughput sequencing data by using JointSLM algorithm. Nucleic Acids Res. 2011, 39, e65. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, L.; Qing, Y.; Li, R.; Li, C.; Li, S.C. SCYN: Single cell CNV profiling method using dynamic programming. BMC Genom. 2021, 22, 651. [Google Scholar] [CrossRef]
- Brouard, J.S.; Bissonnette, N. Variant Calling from RNA-seq Data Using the GATK Joint Genotyping Workflow. Methods Mol. Biol. 2022, 2493, 205–233. [Google Scholar] [CrossRef]
- Patil, G.B.; Lakhssassi, N.; Wan, J.; Song, L.; Zhou, Z.; Klepadlo, M.; Vuong, T.D.; Stec, A.O.; Kahil, S.S.; Colantonio, V.; et al. Whole-genome re-sequencing reveals the impact of the interaction of copy number variants of the rhg1 and Rhg4 genes on broad-based resistance to soybean cyst nematode. Plant Biotechnol. J. 2019, 17, 1595–1611. [Google Scholar] [CrossRef]
- Bellos, E.; Johnson, M.R.; Coin, L.J. cnvHiTSeq: Integrative models for high-resolution copy number variation detection and genotyping using population sequencing data. Genome Biol. 2012, 13, R120. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Getz, G.; Jaffe, D.B.; O’Kelly, M.J.; Zhao, X.; Carter, S.L.; Russ, C.; Nusbaum, C.; Meyerson, M.; Lander, E.S. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods 2009, 6, 99–103. [Google Scholar] [CrossRef]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061–1067. [Google Scholar] [CrossRef]
- Xie, C.; Tammi, M.T. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinform. 2009, 10, 80. [Google Scholar] [CrossRef]
- Yoon, S.; Xuan, Z.; Makarov, V.; Ye, K.; Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009, 19, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Luquette, L.J.; Xi, R.; Park, P.J. rSW-seq: Algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinform. 2010, 11, 432. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; McIntyre, R.E.; Adams, D.J.; Durbin, R. Copy number variant detection in inbred strains from short read sequence data. Bioinformatics 2010, 26, 565–567. [Google Scholar] [CrossRef]
- Zhu, M.; Need, A.C.; Han, Y.; Ge, D.; Maia, J.M.; Zhu, Q.; Heinzen, E.L.; Cirulli, E.T.; Pelak, K.; He, M.; et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am. J. Hum. Genet. 2012, 91, 408–421. [Google Scholar] [CrossRef]
- Sindi, S.S.; Onal, S.; Peng, L.C.; Wu, H.T.; Raphael, B.J. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol. 2012, 13, R22. [Google Scholar] [CrossRef]
- Marschall, T.; Hajirasouliha, I.; Schönhuth, A. MATE-CLEVER: Mendelian-inheritance-aware discovery and genotyping of midsize and long indels. Bioinformatics 2013, 29, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.N.; Sarangi, V.; Moore, R.; Baheti, S.; Bhavsar, J.D.; Couch, F.J.; Kocher, J.P. SoftSearch: Integration of multiple sequence features to identify breakpoints of structural variations. PLoS ONE 2013, 8, e83356. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Merriman, T.R.; Black, M.A. The CNVrd2 package: Measurement of copy number at complex loci using high-throughput sequencing data. Front. Genet. 2014, 5, 248. [Google Scholar] [CrossRef]
- Chu, C.; Zhang, J.; Wu, Y. GINDEL: Accurate genotype calling of insertions and deletions from low coverage population sequence reads. PLoS ONE 2014, 9, e113324. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Xie, W.; Vogel, I.; Choy, K.W.; Chen, F.; Christensen, R.; Zhang, C.; Ge, H.; Jiang, H.; et al. PSCC: Sensitive and reliable population-scale copy number variation detection method based on low coverage sequencing. PLoS ONE 2014, 9, e85096. [Google Scholar] [CrossRef]
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.R.; Hall, I.M.; Quinlan, A.R. Population-based structural variation discovery with Hydra-Multi. Bioinformatics 2015, 31, 1286–1289. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zheng, Z.; Cai, Y.; Chen, T.; Li, C.; Fu, W.; Jiang, Y. CNVcaller: Highly efficient and widely applicable software for detecting copy number variations in large populations. Gigascience 2017, 6, gix115. [Google Scholar] [CrossRef]
- Heldenbrand, J.R.; Baheti, S.; Bockol, M.A.; Drucker, T.M.; Hart, S.N.; Hudson, M.E.; Iyer, R.K.; Kalmbach, M.T.; Kendig, K.I.; Klee, E.W.; et al. Recommendations for performance optimizations when using GATK3.8 and GATK4. BMC Bioinform. 2019, 20, 557. [Google Scholar] [CrossRef]
- Wei, Y.C.; Huang, G.H. CONY: A Bayesian procedure for detecting copy number variations from sequencing read depths. Sci. Rep. 2020, 10, 10493. [Google Scholar] [CrossRef] [PubMed]
- Chanwigoon, S.; Piwluang, S.; Wichadakul, D. inCNV: An Integrated Analysis Tool for Copy Number Variation on Whole Exome Sequencing. Evol. Bioinform. Online 2020, 16, 1176934320956577. [Google Scholar] [CrossRef]
- Knaus, B.J.; Tabima, J.F.; Shakya, S.K.; Judelson, H.S.; Grünwald, N.J. Genome-Wide Increased Copy Number is Associated with Emergence of Dominant Clones of the Irish Potato Famine Pathogen Phytophthora infestans. mBio 2020, 11, e00326-20. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, Y.; Zheng, J.; Wen, Y.; Qu, M.; Kang, S.; Wu, S.; Deng, X.; Hong, K.; Li, S.; et al. A genome-wide survey of copy number variations reveals an asymmetric evolution of duplicated genes in rice. BMC Biol. 2020, 18, 73. [Google Scholar] [CrossRef]
- Juery, C.; Concia, L.; De Oliveira, R.; Papon, N.; Ramírez-González, R.; Benhamed, M.; Uauy, C.; Choulet, F.; Paux, E. New insights into homoeologous copy number variations in the hexaploid wheat genome. Plant Genome 2021, 14, e20069. [Google Scholar] [CrossRef]
- Li, Z.; Han, Y.; Niu, H.; Wang, Y.; Jiang, B.; Weng, Y. Gynoecy instability in cucumber (Cucumis sativus L.) is due to unequal crossover at the copy number variation-dependent Femaleness (F) locus. Hortic. Res. 2020, 7, 32. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, X.; Liu, J.; Mao, C.; Chen, S.; Zhang, Y.; Leng, L. Identification of copy number variation and population analysis of the sacred lotus (Nelumbo nucifera). Biosci. Biotechnol. Biochem. 2020, 84, 2037–2044. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161.e123. [Google Scholar] [CrossRef]
- Ji, G.; Liang, C.; Cai, Y.; Pan, Z.; Meng, Z.; Li, Y.; Jia, Y.; Miao, Y.; Pei, X.; Gong, W.; et al. A copy number variant at the HPDA-D12 locus confers compact plant architecture in cotton. New Phytol. 2021, 229, 2091–2103. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ramasamy, S.; Singh, P.; Hagel, J.M.; Dunemann, S.M.; Chen, X.; Chen, R.; Yu, L.; Tucker, J.E.; Facchini, P.J.; et al. Gene clustering and copy number variation in alkaloid metabolic pathways of opium poppy. Nat. Commun. 2020, 11, 1190. [Google Scholar] [CrossRef]
- Li, J.; Yuan, D.; Wang, P.; Wang, Q.; Sun, M.; Liu, Z.; Si, H.; Xu, Z.; Ma, Y.; Zhang, B.; et al. Cotton pan-genome retrieves the lost sequences and genes during domestication and selection. Genome Biol. 2021, 22, 119. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Chae, G.Y.; Oh, S.; Kim, J.; Mang, H.; Kim, S.; Choi, D. Comparative analysis of de novo genomes reveals dynamic intra-species divergence of NLRs in pepper. BMC Plant Biol. 2021, 21, 247. [Google Scholar] [CrossRef]
- Boatwright, J.L.; Sapkota, S.; Jin, H.; Schnable, J.C.; Brenton, Z.; Boyles, R.; Kresovich, S. Sorghum Association Panel whole-genome sequencing establishes cornerstone resource for dissecting genomic diversity. Plant J. 2022, 111, 888–904. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadian, A.; Yuan, Y.; Bayer, P.E.; Petereit, J.; Severn-Ellis, A.; Tirnaz, S.; Patel, D.; Edwards, D.; Batley, J. Copy Number Variation among Resistance Genes Analogues in Brassica napus. Genes 2022, 13, 2037. [Google Scholar] [CrossRef]
- Bosman, R.N.; Vervalle, J.A.; November, D.L.; Burger, P.; Lashbrooke, J.G. Grapevine genome analysis demonstrates the role of gene copy number variation in the formation of monoterpenes. Front. Plant Sci. 2023, 14, 1112214. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, W.; Zhang, P.; Sun, W.; Han, Y.; Li, L. A comprehensive analysis of copy number variations in diverse apple populations. BMC Genom. 2023, 24, 256. [Google Scholar] [CrossRef]
- Yuan, P.; Huang, P.C.; Martin, T.K.; Chappell, T.M.; Kolomiets, M.V. Duplicated Copy Number Variant of the Maize 9-Lipoxygenase ZmLOX5 Improves 9,10-KODA-Mediated Resistance to Fall Armyworms. Genes 2024, 15, 401. [Google Scholar] [CrossRef]
- Díaz, A.; Zikhali, M.; Turner, A.S.; Isaac, P.; Laurie, D.A. Copy number variation affecting the Photoperiod-B1 and Vernalization-A1 genes is associated with altered flowering time in wheat (Triticum aestivum). PLoS ONE 2012, 7, e33234. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, Y.; Li, X.; Wang, H.; Ding, Y.; Kang, C.; Sun, M.; Li, F.; Wang, J.; Deng, Y.; et al. High frequency DNA rearrangement at qγ27 creates a novel allele for Quality Protein Maize breeding. Commun. Biol. 2019, 2, 460. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, J.; Zhu, Y.; Mo, Y.; Yao, X.F.; Wang, R.; Ku, W.; Huang, Z.; Xia, S.; Tong, J.; et al. The Copy Number Variation of OsMTD1 Regulates Rice Plant Architecture. Front. Plant Sci. 2020, 11, 620282. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.W.; Wong, F.L.; Luk, C.Y.; Chung, C.Y.; Yung, W.S.; Wang, Z.; Xie, M.; Song, S.; Chung, G.; et al. Increased copy number of gibberellin 2-oxidase 8 genes reduced trailing growth and shoot length during soybean domestication. Plant J. 2021, 107, 1739–1755. [Google Scholar] [CrossRef]
- Cardone, M.F.; D’Addabbo, P.; Alkan, C.; Bergamini, C.; Catacchio, C.R.; Anaclerio, F.; Chiatante, G.; Marra, A.; Giannuzzi, G.; Perniola, R.; et al. Inter-varietal structural variation in grapevine genomes. Plant J. 2016, 88, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.M.; Song, C.; Bradbury, P.J.; Costich, D.; de Leon, N.; Doebley, J.; Elshire, R.J.; Gaut, B.; Geller, L.; Glaubitz, J.C.; et al. Maize HapMap2 identifies extant variation from a genome in flux. Nat. Genet. 2012, 44, 803–807. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, F.; Zhang, J.; Liu, H.; Rahman, S.; Islam, S.; Ma, W.; She, M. Application of CRISPR/Cas9 in Crop Quality Improvement. Int. J. Mol. Sci. 2021, 22, 4206. [Google Scholar] [CrossRef]
- Mareri, L.; Milc, J.; Laviano, L.; Buti, M.; Vautrin, S.; Cauet, S.; Mascagni, F.; Natali, L.; Cavallini, A.; Bergès, H.; et al. Influence of CNV on transcript levels of HvCBF genes at Fr-H2 locus revealed by resequencing in resistant barley cv. ‘Nure’ and expression analysis. Plant Sci. 2020, 290, 110305. [Google Scholar] [CrossRef]
- Saxena, R.K.; Edwards, D.; Varshney, R.K. Structural variations in plant genomes. Brief. Funct. Genom. 2014, 13, 296–307. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, Y.; Kuang, H.; Chen, J. Frequent loss of lineages and deficient duplications accounted for low copy number of disease resistance genes in Cucurbitaceae. BMC Genom. 2013, 14, 335. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Lye, Z.N.; Purugganan, M.D. Copy Number Variation in Domestication. Trends Plant Sci. 2019, 24, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wang, C.; Xu, Q.; Feng, Y.; Yuan, X.; Yu, H.; Wang, Y.; Tang, S.; Wei, X. Detection of copy number variations in rice using array-based comparative genomic hybridization. BMC Genom. 2011, 12, 372. [Google Scholar] [CrossRef]
- Swanson-Wagner, R.A.; Eichten, S.R.; Kumari, S.; Tiffin, P.; Stein, J.C.; Ware, D.; Springer, N.M. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010, 20, 1689–1699. [Google Scholar] [CrossRef]
- Wang, Y.X.; Xiong, G.S.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.X.; Zeng, L.J.; Xu, E.B.; Xu, J.; et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef]
- Choi, J.Y.; Zaidem, M.; Gutaker, R.; Dorph, K.; Singh, R.K.; Purugganan, M.D. The complex geography of domestication of the African rice Oryza glaberrima. PLoS Genet. 2019, 15, e1007414. [Google Scholar] [CrossRef]
- McHale, L.K.; Haun, W.J.; Xu, W.W.; Bhaskar, P.B.; Anderson, J.E.; Hyten, D.L.; Gerhardt, D.J.; Jeddeloh, J.A.; Stupar, R.M. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 2012, 159, 1295–1308. [Google Scholar] [CrossRef]
- Lin, Z.; Li, X.; Shannon, L.M.; Yeh, C.T.; Wang, M.L.; Bai, G.; Peng, Z.; Li, J.; Trick, H.N.; Clemente, T.E.; et al. Parallel domestication of the Shattering1 genes in cereals. Nat. Genet. 2012, 44, 720–724. [Google Scholar] [CrossRef]
- Dar, A.M.; Touseef, H.; Nawaz, K.; Khan, Y.; Sahu, P.P. Editorial: Genomics in plant sciences: Understanding and development of stress-tolerant plants. Front. Plant Sci. 2023, 14, 1222818. [Google Scholar] [CrossRef]
- Aziz, M.A.; Masmoudi, K. Molecular Breakthroughs in Modern Plant Breeding Techniques. Hortic. Plant J. 2024, 11, 15–41. [Google Scholar] [CrossRef]
- Lee, T.G.; Kumar, I.; Diers, B.W.; Hudson, M.E. Evolution and selection of Rhg1, a copy-number variant nematode-resistance locus. Mol. Ecol. 2015, 24, 1774–1791. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.E.; Lee, T.G.; Guo, X.; Melito, S.; Wang, K.; Bayless, A.M.; Wang, J.; Hughes, T.J.; Willis, D.K.; Clemente, T.E.; et al. Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 2012, 338, 1206–1209. [Google Scholar] [CrossRef]
- Xu, X.; Liu, X.; Ge, S.; Jensen, J.D.; Hu, F.; Li, X.; Dong, Y.; Gutenkunst, R.N.; Fang, L.; Huang, L.; et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 2011, 30, 105–111. [Google Scholar] [CrossRef]
- Lu, P.; Han, X.; Qi, J.; Yang, J.; Wijeratne, A.J.; Li, T.; Ma, H. Analysis of Arabidopsis genome-wide variations before and after meiosis and meiotic recombination by resequencing Landsberg erecta and all four products of a single meiosis. Genome Res. 2012, 22, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Boocock, J.; Chagné, D.; Merriman, T.R.; Black, M.A. The distribution and impact of common copy-number variation in the genome of the domesticated apple, Malus x domestica Borkh. BMC Genom. 2015, 16, 848. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Leal-Bertioli, S.C.; Lion, M.B.; Santos, V.L.; Pappas, G., Jr.; Cannon, S.B.; Guimarães, P.M. A large scale analysis of resistance gene homologues in Arachis. Mol. Genet. Genom. 2003, 270, 34–45. [Google Scholar] [CrossRef]
- Zhai, J.; Jeong, D.H.; De Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; González, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes. Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef]
- Ionita-Laza, I.; Rogers, A.J.; Lange, C.; Raby, B.A.; Lee, C. Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis. Genomics 2009, 93, 22–26. [Google Scholar] [CrossRef]
- Drackley, A.; Brew, C.; Wlodaver, A.; Spencer, S.; Leuer, K.; Rathbun, P.; Charrow, J.; Wieneke, X.; Yap, K.L.; Ing, A. Utility and Outcomes of the 2019 American College of Medical Genetics and Genomics-Clinical Genome Resource Guidelines for Interpretation of Copy Number Variants with Borderline Classifications at an Academic Clinical Diagnostic Laboratory. J. Mol. Diagn. 2022, 24, 1100–1111. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, U.; Li, X.; Fan, Y.; Chang, W.; Niu, Y.; Li, J.; Qu, C.; Lu, K. Multi-omics revolution to promote plant breeding efficiency. Front Plant Sci. 2022, 13, 1062952. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, Z.; Sun, X. Single-cell and spatial multi-omics in the plant sciences: Technical advances, applications, and perspectives. Plant Commun. 2023, 4, 100508. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.; Unckless, R.L. A Deep Learning Approach for Detecting Copy Number Variation in Next-Generation Sequencing Data. G3 Genes. Genomes Genet. 2019, 9, 3575–3582. [Google Scholar] [CrossRef]
- Chen, L.; Liu, G.; Zhang, T. Integrating machine learning and genome editing for crop improvement. aBIOTECH 2024, 5, 262–277. [Google Scholar] [CrossRef]
| Category | Publication Year | No. of Samples | CNV Detection Tools | References |
|---|---|---|---|---|
| Glycine max | 2019 | 106 | GATK, SAMTools | [86] |
| Potato | 2019 | 47 | GATK | [107] |
| Arabidopsis thaliana | 2020 | 1060 | CNVnator | [34] |
| Rice | 2020 | 93 | CNVnator, Delly, CtgRefCNV | [108] |
| hexaploid wheat | 2020 | 16 | GMAP | [109] |
| Barley | 2020 | 397 | ExomeDepth | [81] |
| Cucumber | 2020 | 9 | FISH qPCR | [110] |
| Lotus | 2020 | 24 | Delly, Manta | [111] |
| Tomato | 2020 | 100 | SVCollector | [112] |
| Cotton | 2020 | 2464 | GATK | [113] |
| Poppy | 2020 | 10 | CNVnator | [114] |
| Cotton | 2021 | 1961 | CNVcaller | [115] |
| Chili Pepper | 2021 | 2 | Illumina HiSeq X-ten, NovaSeq 6000 | [116] |
| Sorghum | 2022 | 400 | GATK | [117] |
| Brassica napus | 2022 | 8 | CNVnator | [118] |
| Grape | 2023 | 82 | 2−DDCt method, QuantStudio | [119] |
| Apple | 2023 | 346 | SpeedSeq, Lumpy, CNVnator | [120] |
| Zea mays | 2024 | 6 | ddPCR | [121] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silaiyiman, S.; Liu, J.; Wu, J.; Ouyang, L.; Cao, Z.; Shen, C. A Systematic Review of the Advances and New Insights into Copy Number Variations in Plant Genomes. Plants 2025, 14, 1399. https://doi.org/10.3390/plants14091399
Silaiyiman S, Liu J, Wu J, Ouyang L, Cao Z, Shen C. A Systematic Review of the Advances and New Insights into Copy Number Variations in Plant Genomes. Plants. 2025; 14(9):1399. https://doi.org/10.3390/plants14091399
Chicago/Turabian StyleSilaiyiman, Saimire, Jiaxuan Liu, Jiaxin Wu, Lejun Ouyang, Zheng Cao, and Chao Shen. 2025. "A Systematic Review of the Advances and New Insights into Copy Number Variations in Plant Genomes" Plants 14, no. 9: 1399. https://doi.org/10.3390/plants14091399
APA StyleSilaiyiman, S., Liu, J., Wu, J., Ouyang, L., Cao, Z., & Shen, C. (2025). A Systematic Review of the Advances and New Insights into Copy Number Variations in Plant Genomes. Plants, 14(9), 1399. https://doi.org/10.3390/plants14091399