
Transcriptome and Small-RNA Sequencing Reveals the Response Mechanism of Brassica napus to Waterlogging Stress


Abstract
1. Introduction
2. Results
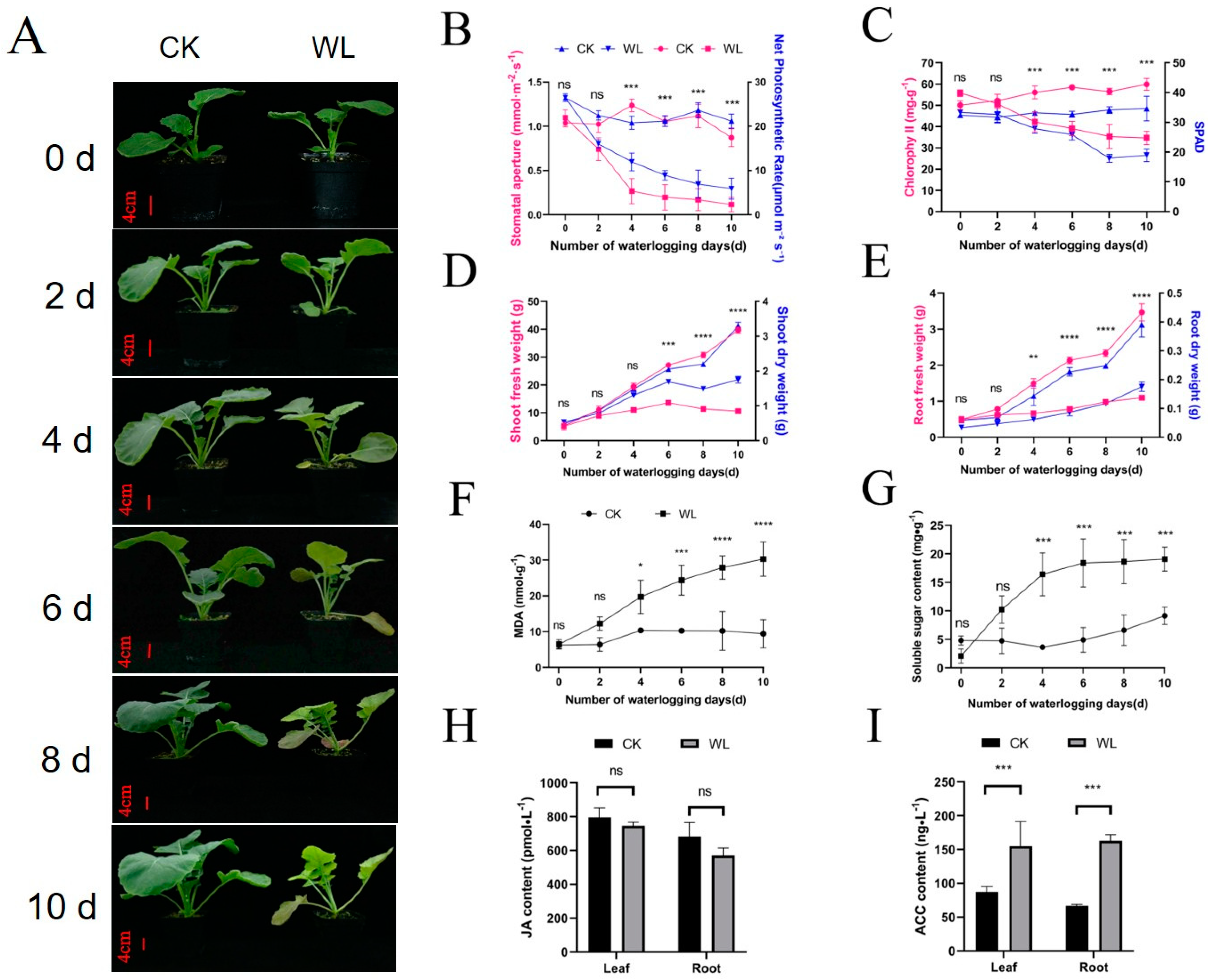
2.1. Waterlogging Inhibits Growth and Alters Physiology of Rapeseed
2.2. DEGs Identified in Leaves and Roots Under Waterlogging Stress
2.3. Small RNAs Identified in Leaves and Roots Under Waterlogging Stress
2.4. Annotation of miRNA Families
2.5. Differentially Expressed miRNAs Under Waterlogging Stress in Rapeseed
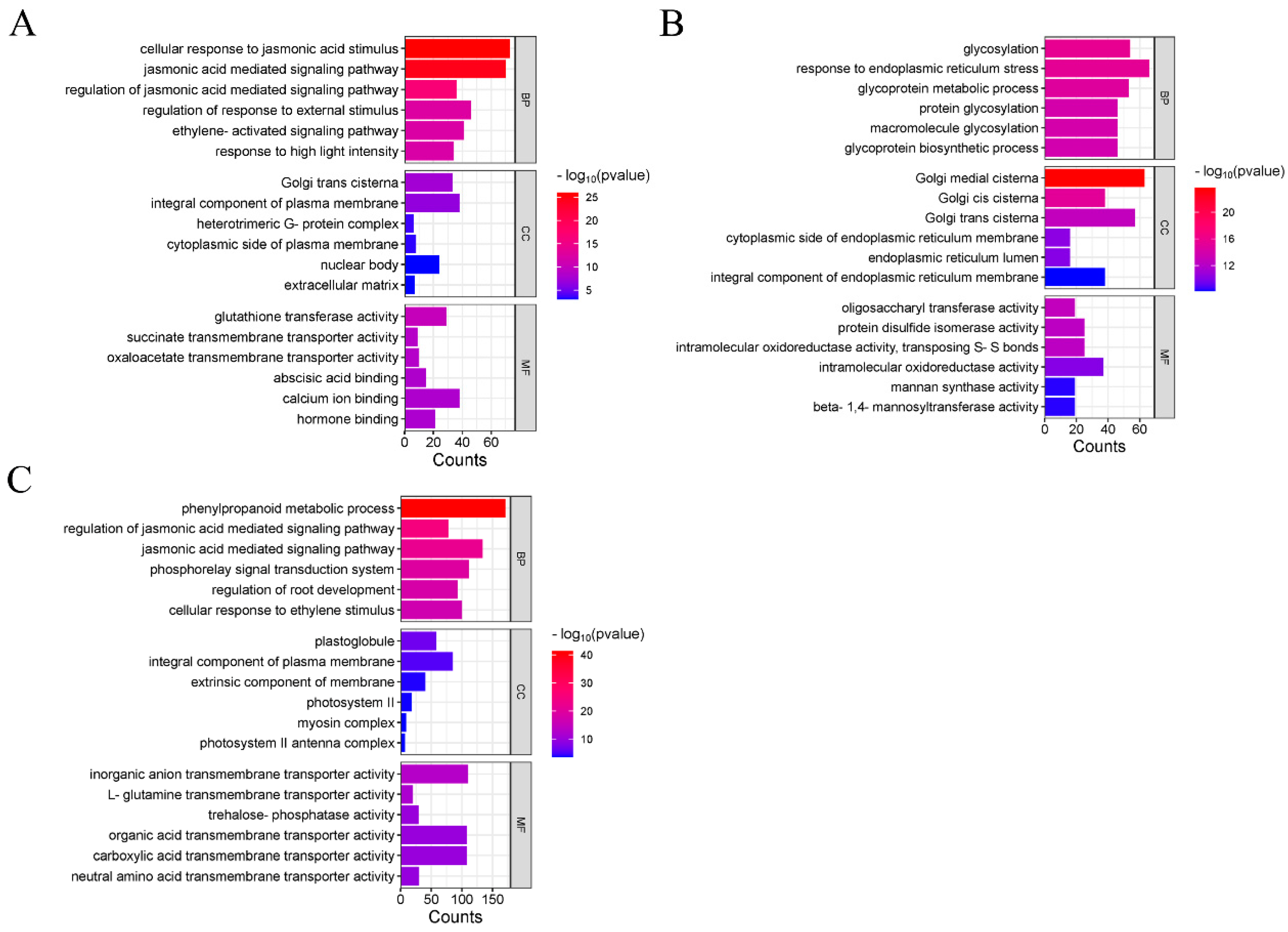
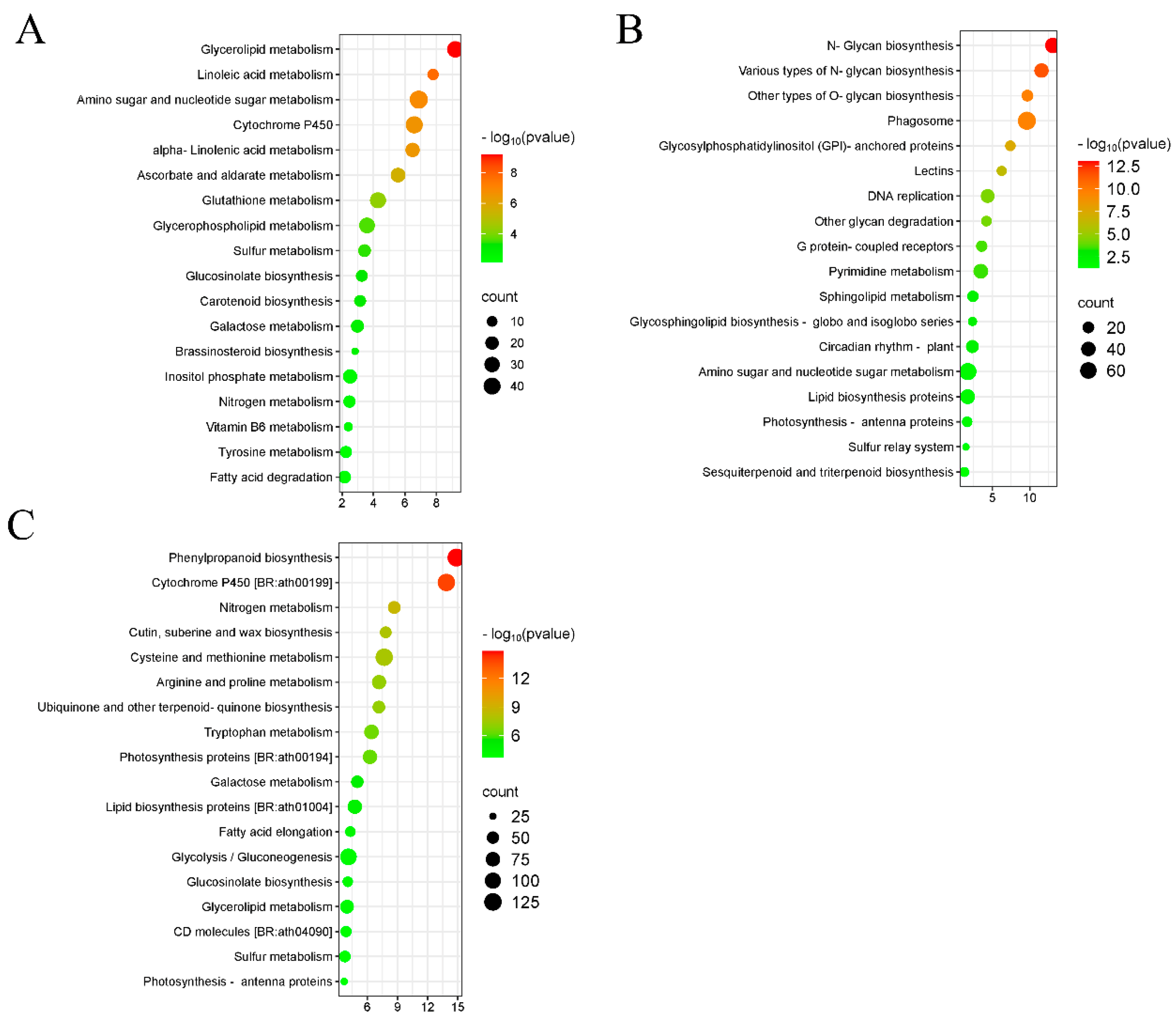
2.6. GO and KEGG Analysis of DEmiRNA Target Genes
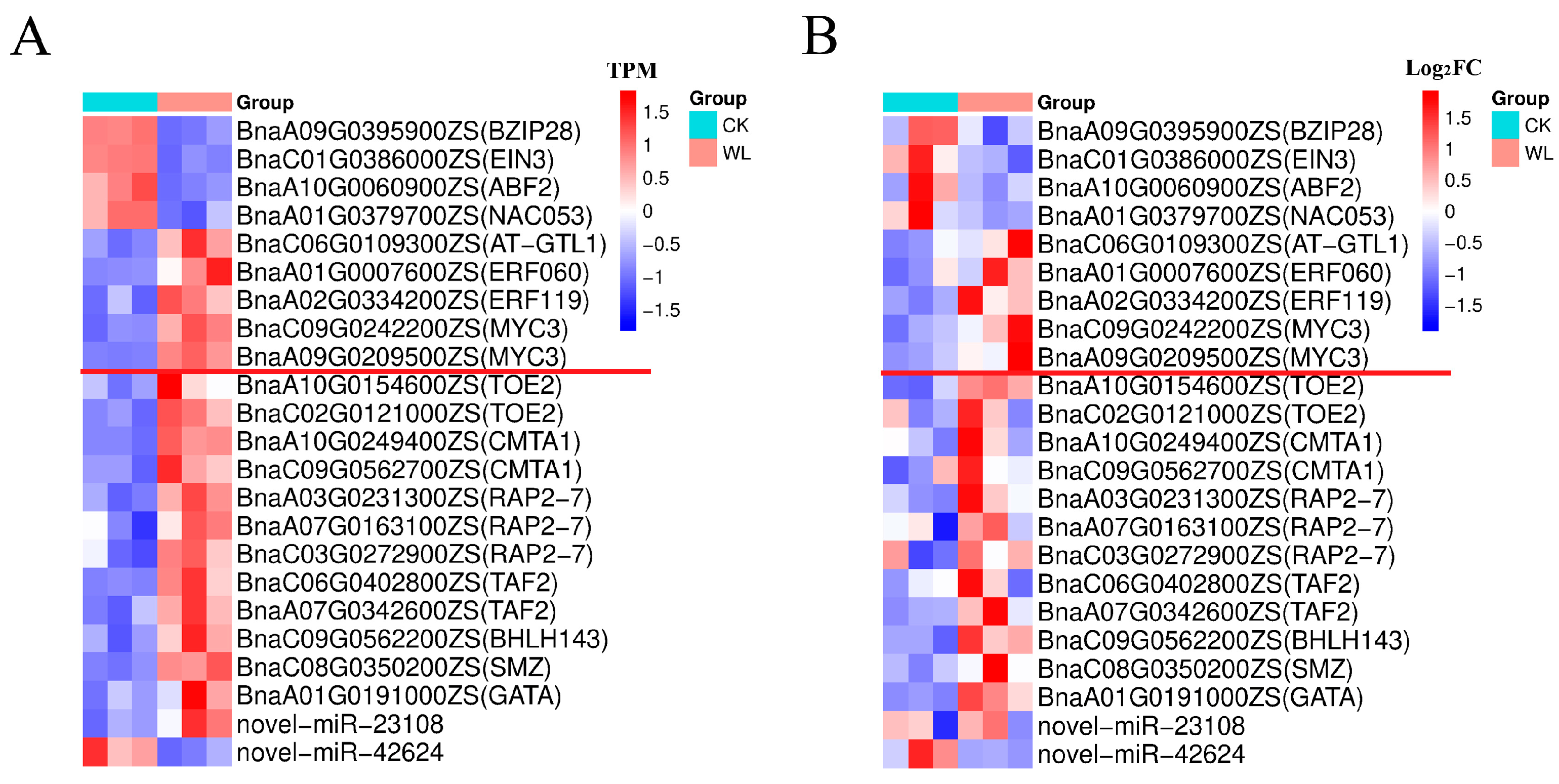
2.7. Transcription Factors in the DEmiRNAs
3. Discussion
4. Materials and Methods
4.1. Materials and Waterlogging Treatment
4.2. Measurement of Physiological and Biochemical Indices
4.3. Transcriptome Data Analysis
4.4. Small RNA Data Analysis
4.5. miRNA Classification and Target Gene Prediction
4.6. GO and KEGG Analysis
4.7. Real-Time Quantitative PCR Analysis
4.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manghwar, H.K.; Hussain, A.; Alam, I. Waterlogging stress in plants: Unraveling the mechanisms and impacts on growth, development, and productivity. Environ. Exp. Bot. 2024, 224, 105824. [Google Scholar] [CrossRef]
- Cheng, H.T.; Hao, M.Y.; Wang, W.X. Integrative RNA- and miRNA-profile analysis reveals a likely role of BR and Auxin signaling in branch angle regulation of B. napus. Int. J. Mol. Sci. 2017, 18, 887. [Google Scholar] [CrossRef]
- Huang, J.D.; Cao, X.Y.; Kuai, J. Evaluation of production capacity for rice-rapeseed cropping system in China. Field Crops Res. 2023, 293, 108842. [Google Scholar] [CrossRef]
- Fukao, T.; Barrera-Figueroa, B.E.; Juntawong, P. Submergence and waterlogging stress in plants: A review highlighting research opportunities and understudied aspects. Front. Plant Sci. 2019, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Boem, F.H.G.; Lavado, R.S.; Porcelli, C.A. Note on the effects of winter and spring waterlogging on growth, chemical composition and yield of rapeseed. Field Crops Res. 1996, 47, 175–179. [Google Scholar] [CrossRef]
- Zou, X.L.; Tan, X.Y.; Hu, C.W. The transcriptome of B. napus L. roots under waterlogging at the seedling stage. Int. J. Mol. Sci. 2013, 14, 2637–2651. [Google Scholar] [CrossRef]
- Hong, B.; Zhou, B.Q.; Peng, Z.C. Tissue-specific transcriptome and metabolome analysis reveals the response mechanism of B. napus to waterlogging stress. Int. J. Mol. Sci. 2023, 24, 6015. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y.; Chen, Y. The roles of cell wall polysaccharides in response to waterlogging stress in Brassica napus L. root. BMC Biol. 2024, 22, 191. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, J.; Kuang, L. Effects of waterlogging stress on early seedling development and transcriptomic responses in Brassica napus. Mol. Breed. 2020, 40, 85. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, K.S.; Jang, Y.S. Global gene expression responses to waterlogging in leaves of rape seedlings. Plant Cell Rep. 2014, 33, 289–299. [Google Scholar] [CrossRef]
- Pan, J.W.; Sharif, R.; Xu, X.W. Mechanisms of waterlogging tolerance in plants: Research progress and prospects. Front. Plant Sci. 2021, 11, 627331. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Zhang, H.; Chen, W.X. The role of ethylene in the regulation of plant response mechanisms to waterlogging stress. Plant Cell Rep. 2024, 43, 278. [Google Scholar] [CrossRef]
- Kim, Y.H.; Seo, C.W.; Khan, A.L. Exo-ethylene application mitigates waterlogging stress in soybean (Glycine max L.). BMC Plant biology 2018, 18, 254. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.H.; Li, Q.Q.; Ma, X.T. Waterlogging-induced adventitious root formation in cucumber is regulated by ethylene and auxin through reactive oxygen species signalling. Plant Cell Environ. 2019, 42, 1458–1470. [Google Scholar] [CrossRef]
- Pan, J.W.; Sohail, H.; Sharif, R. Cucumber JASMONATE ZIM-DOMAIN 8 interaction with transcription factor MYB6 impairs waterlogging-triggered adventitious rooting. Plant Physiol. 2025, 197, kiae351. [Google Scholar] [CrossRef]
- Hudgins, J.W.; Franceschi, V.R. Methyl Jasmonate-induced Ethylene production is responsible for conifer phloem defense responses and reprogramming of stem cambial zone for traumatic resin duct formation. Plant Physiol. 2004, 135, 2134–2149. [Google Scholar] [CrossRef]
- Zhang, B.H. MicroRNA: A new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.M.F.; Klein, D.; Cubela, S.A. The role of microRNAs in diabetes-related oxidative stress. Int. J. Mol. Sci. 2019, 20, 5423. [Google Scholar] [CrossRef]
- Liu, D.M.; Song, Y.; Chen, Z.X. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef]
- Sharma, R.; Upadhyay, S.; Bhat, B. Abiotic stress induced miRNA-TF-gene regulatory network: A structural perspective. Genomics 2020, 112, 412–422. [Google Scholar] [CrossRef]
- Ma, Z.M.; Hu, L.J. MicroRNA: A dynamic player from signalling to abiotic tolerance in plants. Int. J. Mol. Sci. 2023, 24, 11364. [Google Scholar] [CrossRef]
- Shi, G.Q.; Fu, J.Y.; Rong, L.J. TaMIR1119, a miRNA family member of wheat (Triticum aestivum), is essential in the regulation of plant drought tolerance. J. Integr. Agric. 2018, 17, 2369–2378. [Google Scholar] [CrossRef]
- Ding, Y.H.; Ma, Y.Z.; Liu, N. microRNAs involved in auxin signalling modulate male sterility under high-temperature stress in cotton (Gossypium hirsutum). Plant J. 2017, 91, 977–994. [Google Scholar] [CrossRef]
- Gao, P.; Bai, X.; Yang, L. Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 2010, 231, 991–1001. [Google Scholar] [CrossRef]
- Moro, B.; Chorostecki, U.; Arikit, S. Efficiency and precision of microRNA biogenesis modes in plants. Nucleic Acids Res. 2018, 46, 10709–10723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Hegarty, J.S.; Padilla, M. Manipulation of the microRNA172–AP2L2 interaction provides precise control of wheat and triticale plant height. Plant Biotechnol. J. 2025, 23, 333–335. [Google Scholar] [CrossRef]
- Sun, X.; Wang, M.Q.; Leng, X.P. Characterization of the regulation mechanism of grapevine microRNA172 family members during flower development. BMC Plant Biol. 2020, 20, 409. [Google Scholar] [CrossRef] [PubMed]
- Frazier, T.P.; Sun, G.; Burklew, C.E. Salt and drought stresses induce the aberrant expression of microRNA genes in Tobacco. Mol. Biotechnol. 2011, 49, 159–165. [Google Scholar] [CrossRef]
- Luan, M.D.; Xu, M.Y.; Lu, Y.M. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 2015, 555, 178–185. [Google Scholar] [CrossRef]
- Lynch, J.P. Root phenes that reduce the metabolic costs of soil exploration: Opportunities for 21st century agriculture. Plant Cell Environ. 2015, 38, 1775–1784. [Google Scholar] [CrossRef]
- Ashraf, M.A. Waterlogging stress in plants: A review. Afr. J. Agric. Res. 2012, 7, 1976–1981. [Google Scholar] [CrossRef]
- Liu, M.; Zwiazek, J.J. Oxidative stress impedes recovery of canola (Brassica napus) plants from waterlogging by inhibiting aquaporin-mediated root water transport. Environ. Exp. Bot. 2022, 200, 104931. [Google Scholar] [CrossRef]
- Li, J.; Iqbal, S.; Zhang, Y. Transcriptome analysis reveals genes of flooding-tolerant and flooding-sensitive rapeseeds differentially respond to fooding at the germination stage. Plants 2021, 10, 693. [Google Scholar] [CrossRef]
- Zheng, L.J.; Zhang, X.G.; Zhang, H.J. The miR164-dependent regulatory pathway in developing maize seed. Mol. Genet. Genom. MGG 2019, 294, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.J.; Zhu, H.T.; Li, N. The miR393-target module regulates plant development and responses to biotic and abiotic stresses. Int. J. Mol. Sci. 2022, 23, 9477. [Google Scholar] [CrossRef]
- Bernardi, Y.; Ponso, M.A.; Belén, F. MicroRNA miR394 regulates flowering time in Arabidopsis thaliana. Plant Cell Rep. 2022, 41, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Hao, M.Y.; Wang, L. The cotton miR477-CBP60A module participates in plant defense against Verticillium dahlia. Mol. Plant-Microbe Interact. MPMI 2020, 33, 624–636. [Google Scholar] [CrossRef]
- Wu, B.F.; Li, W.F.; Xu, H.Y. Role of cin-miR2118 in drought stress responses in Caragana intermedia and Tobacco. Gene 2015, 574, 34–40. [Google Scholar] [CrossRef]
- Zeng, Z.C.; Li, Y.L.; Pan, Y.J. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef]
- Jiang, M.Y.; Jike, Y.J.; Liu, K.C. Exosome-mediated miR-144-3p promotes ferroptosis to inhibit osteosarcoma proliferation, migration, and invasion through regulating ZEB1. Mol. Cancer 2023, 22, 113. [Google Scholar] [CrossRef]
- Ghabooli, M.; Beheshti Rooy, S.S.; Mohseni Fard, E. miR395 is involved in response to cold stress and modulation of sulfate and phosphate deficiency in Grape (Vitis vinifera). J. Plant Mol. Breed. 2019, 7, 56–66. [Google Scholar] [CrossRef]
- Pegler, J.L.; Oultram, J.M.J.; Grof, C.P.L. Molecular manipulation of the miR399/PHO2 expression module alters the salt stress response of Arabidopsis thaliana. Plants 2020, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Sang, Q.; Vayssières, A.; Ó’Maoiléidigh, D.S. MicroRNA172 controls inflorescence meristem size through regulation of APETALA2 in Arabidopsis. New Phytol. 2022, 235, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.T.; Kuo, Y.W.; King, Y.C. Regulation of micoRNA2111 and its target IbFBK in sweet potato on wounding. Plant Sci. 2020, 292, 110391. [Google Scholar] [CrossRef]
- Ma, J.; Du, G.; Li, X. A major locus controlling malondialdehyde content under water stress is associated with fusarium crown rot resistance in wheat. Mol. Genet. Genom. 2015, 290, 1955–1962. [Google Scholar] [CrossRef]
- Qi, X.; Li, Q.; Shen, J. Sugar enhances waterlogging-induced adventitious root formation in cucumber by promoting auxin transport and signalling. Plant Cell Environ. 2020, 43, 1545–1557. [Google Scholar] [CrossRef]
- Ma, S.; Gai, P.; Geng, B. Exogenous melatonin improves waterlogging tolerance in wheat through promoting antioxidant enzymatic activity and carbon assimilation. Agronomy 2022, 12, 2876. [Google Scholar] [CrossRef]
- Ksouri, N.; Jiménez, S.; Wells, C.E. Transcriptional responses in root and leaf of Prunus persica under drought stress using RNA sequencing. Front. Plant Sci. 2016, 7, 1715. [Google Scholar] [CrossRef]
- Christianson, J.A.; Llewellyn, D.J.; Dennis, E.S. Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypium hirsutum L.). Plant Cell Physiol. 2010, 51, 21–37. [Google Scholar] [CrossRef]
- Arora, K.; Panda, K.K.; Mittal, S. RNAseq revealed the important gene pathways controlling adaptive mechanisms under waterlogged stress in maize. Sci. Rep. 2017, 7, 10950. [Google Scholar] [CrossRef]
- Yao, Q. Crucial waterlogging-responsive genes and pathways revealed by comparative physiology and transcriptome in tropical and temperate maize (Zea mays L.) inbred lines. J. Plant Biol. 2021, 64, 313–325. [Google Scholar] [CrossRef]
- Ding, L.N.; Liu, R.; Li, T. Physiological and comparative transcriptome analyses reveal the mechanisms underlying waterlogging tolerance in a rapeseed anthocyanin-more mutant. Biotechnol. Biofuels Bioprod. 2022, 15, 55. [Google Scholar] [CrossRef]
- Kumar, J.; Gunapati, S.; Kianian, S.F. Comparative analysis of transcriptome in two wheat genotypes with contrasting levels of drought tolerance. Protoplasma 2018, 255, 1487–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Liu, P.; Qing, C.Y. Comparative transcriptome analyses of maize seedling root responses to salt stress. PeerJ 2021, 9, e10765. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.H.; Liu, Z.J.; Zou, X.L. Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol. Plant. 2013, 147, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.W.; Wang, K.X.; Pan, J.W. Small RNA sequencing identifies cucumber miRNA roles in waterlogging-triggered adventitious root primordia formation. Mol. Biol. Rep. 2019, 46, 6381–6389. [Google Scholar] [CrossRef]
- Kang, Y.C.; Yang, X.Y.; Liu, Y.H. Integration of mRNA and miRNA analysis reveals the molecular mechanism of potato (Solanum tuberosum L.) response to alkali stress. Int. J. Biol. Macromol. 2021, 182, 938–949. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, S.Q.; Xiang, P. Integrated small RNA, transcriptome and physiological approaches provide insight into Taxodium hybrid ‘Zhongshanshan’ roots in acclimation to prolonged flooding. Tree Physiol. 2024, 44, 4. [Google Scholar] [CrossRef]
- Keska, K.; Szcześniak, M.W.; Adamus, A. Waterlogging-stress-responsive LncRNAs, their regulatory relationships with miRNAs and target genes in cucumber (Cucumis sativus L.). Int. J. Mol. Sci. 2021, 22, 8197. [Google Scholar] [CrossRef]
- Yu, F.; Tan, Z.; Fang, T. A Comprehensive transcriptomics analysis reveals long non-coding RNA to be involved in the key metabolic pathway in response to waterlogging stress in maize. Genes 2020, 11, 267. [Google Scholar] [CrossRef]
- Rauf, M.; Arif, M.; Fisahn, J. NAC transcription factor Speedy Hyponastic Growth regulates flooding-induced leaf movement in Arabidopsis. Plant Cell 2013, 25, 4941–4955. [Google Scholar] [CrossRef] [PubMed]
- Bashar, K.K. Hormone dependent survival mechanisms of plants during post-waterlogging stress. Plant Signal. Behav. 2018, 13, e1529522. [Google Scholar] [CrossRef] [PubMed]
- Stinziano, J.R.; Morgan, P.B.; Lynch, D.J. The rapid A-Ci response: Photosynthesis in the phenomic era. Plant Cell Environ. 2017, 40, 1256–1262. [Google Scholar] [CrossRef]
- Fatima, A.; Safdar, N.; Ain, N.U. Abscisic acid-loaded ZnO nanoparticles as drought tolerance inducers in Zea mays L. with physiological and biochemical attributes. J. Plant Growth Regul. 2023, 42, 7280–7293. [Google Scholar] [CrossRef]
- Huang, W.W.; Ratkowsky, D.A.; Hui, C. Leaf fresh weight versus dry weight: Which is better for describing the scaling relationship between leaf biomass and leaf area for broad-leaved plants? Forests 2019, 10, 256. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Wu, H.J.; Ma, Y.K.; Chen, T. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Lilove, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Hussain, M.A.; Huang, Y.; Luo, D. Integrative analyses reveal Bna-miR397a–BnaLAC2 as a potential modulator of low-temperature adaptability in Brassica napus L. Plant Biotechnol. J. 2025; early view. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Chen, X.; Zhang, S.S. The genome sequence archive family: Toward explosive data growth and diverse data types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef] [PubMed]
- CNCB-NGDC Members and Partners. Database resources of the national genomics data center, China national center for bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Read Type | Total | rRNA | snRNA | tRNA | Repeat | Other |
|---|---|---|---|---|---|---|
| Leaf-CKT-1 | 13,884,720 | 7,556,783 (54.43%) | 48,196 (0.35%) | 2,503,157 (18.03%) | 2,709,768 (19.52%) | 3,982,761 (28.68%) |
| Leaf-CKT-2 | 5,711,607 | 3,228,497 (56.53%) | 14,078 (0.25%) | 942,867 (16.51%) | 1,155,668 (20.23%) | 1,616,082 (28.29%) |
| Leaf-CKT-3 | 8,545,681 | 4,984,691 (58.33%) | 39,373 (0.46%) | 1,167,813 (13.67%) | 1,785,472 (20.89%) | 2,484,058 (29.07%) |
| Leaf-WLT-1 | 9,222,335 | 4,444,472 (48.19%) | 60,181 (0.65%) | 1,022,050 (11.08%) | 2,030,677 (22.02%) | 3,743,626 (40.59%) |
| Leaf-WLT-2 | 10,565,031 | 6,220,196 (58.88%) | 60,938 (0.58%) | 854,937 (8.09%) | 2,587,181 (24.49%) | 3,501,670 (33.14%) |
| Leaf-WLT-3 | 32,933,054 | 18,135,516 (55.07%) | 310,346 (0.94%) | 3,369,992 (10.23%) | 8,104,513 (24.61%) | 11,266,420 (34.21%) |
| Root-CKT-1 | 5,556,492 | 2,329,546 (41.92%) | 16,652 (0.3%) | 1,033,337 (18.60%) | 1,392,040 (25.05%) | 2,168,195 (39.02%) |
| Root-CKT-2 | 4,516,520 | 1,701,149 (37.67%) | 15,000 (0.33%) | 879,215 (19.47%) | 1,064,131 (23.56%) | 1,911,908 (42.33%) |
| Root-CKT-3 | 6,060,957 | 2,413,813 (39.83%) | 22,376 (0.37%) | 1,070,520 (17.66%) | 1,416,834 (23.38%) | 2,541,770 (41.94%) |
| Root-WLT-1 | 10,045,342 | 6,463,385 (64.34%) | 25,259 (0.25%) | 1,141,681 (11.37%) | 3,856,248 (38.39%) | 2,400,758 (23.90%) |
| Root-WLT-2 | 13,201,016 | 8,285,263 (62.76%) | 35,380 (0.27%) | 1,473,133 (11.16%) | 5,008,072 (37.94%) | 3,378,301 (25.59%) |
| Root-WLT-3 | 11,927,136 | 7,563,060 (63.41%) | 27,194 (0.23%) | 1,461,914 (12.26%) | 4,629,966 (38.82%) | 2,861,382 (23.99%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Ge, L.; Wang, K.; Wang, N.; Wang, X. Transcriptome and Small-RNA Sequencing Reveals the Response Mechanism of Brassica napus to Waterlogging Stress. Plants 2025, 14, 1340. https://doi.org/10.3390/plants14091340
Song X, Ge L, Wang K, Wang N, Wang X. Transcriptome and Small-RNA Sequencing Reveals the Response Mechanism of Brassica napus to Waterlogging Stress. Plants. 2025; 14(9):1340. https://doi.org/10.3390/plants14091340
Chicago/Turabian StyleSong, Xianshuai, Lan Ge, Kaifeng Wang, Nian Wang, and Xinfa Wang. 2025. "Transcriptome and Small-RNA Sequencing Reveals the Response Mechanism of Brassica napus to Waterlogging Stress" Plants 14, no. 9: 1340. https://doi.org/10.3390/plants14091340
APA StyleSong, X., Ge, L., Wang, K., Wang, N., & Wang, X. (2025). Transcriptome and Small-RNA Sequencing Reveals the Response Mechanism of Brassica napus to Waterlogging Stress. Plants, 14(9), 1340. https://doi.org/10.3390/plants14091340