
Pan-Genome-Based Characterization of the SRS Transcription Factor Family in Foxtail Millet
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
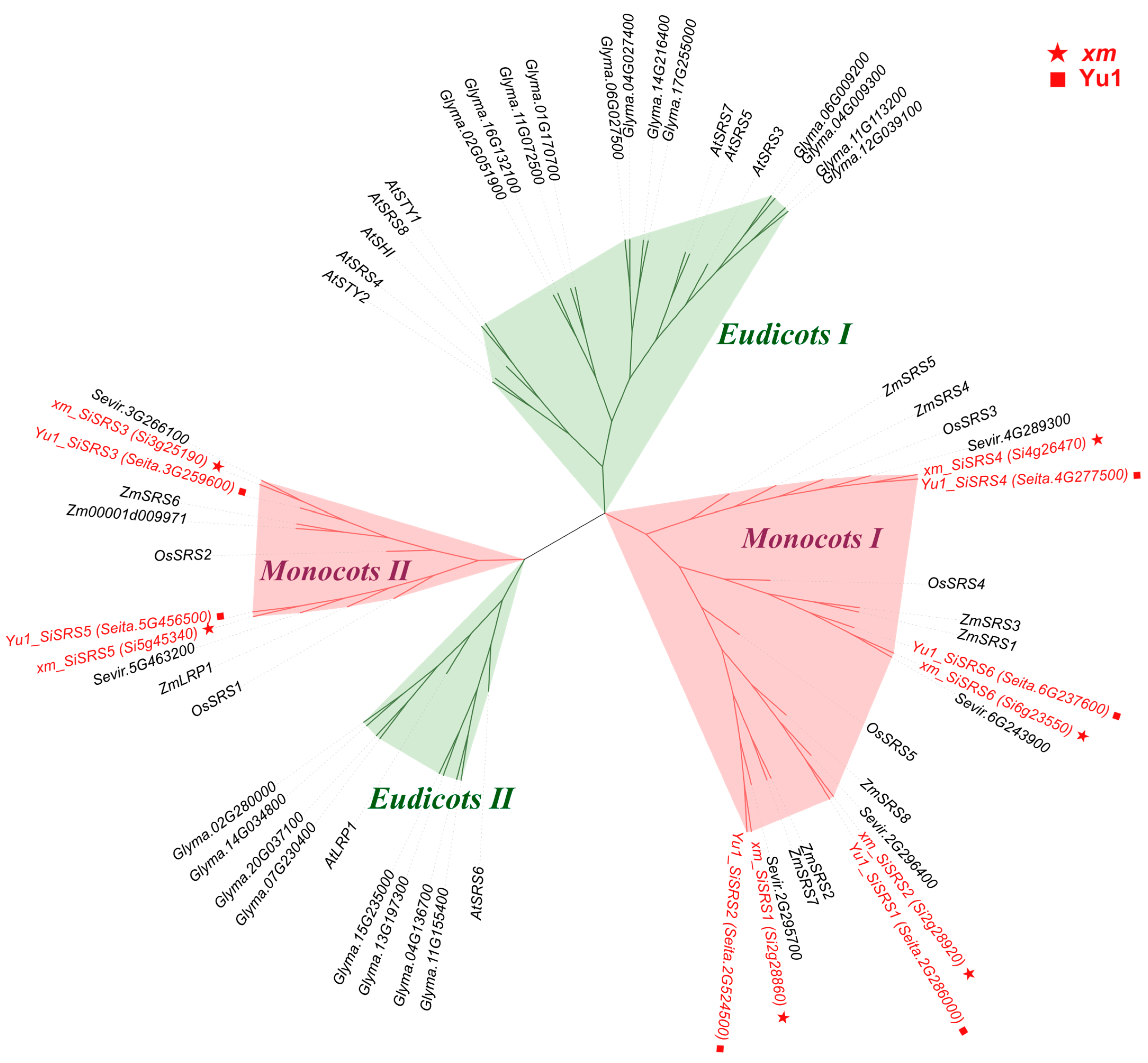
2.2. Identification and Phylogenetic Analysis of SRS Family Members
2.3. Analysis of Promoter Sequences of SRS Family Members at the Pan-Genome Level
2.4. Motif and Conserved Domain Analysis
2.5. Collinearity and Selective Pressure Analysis
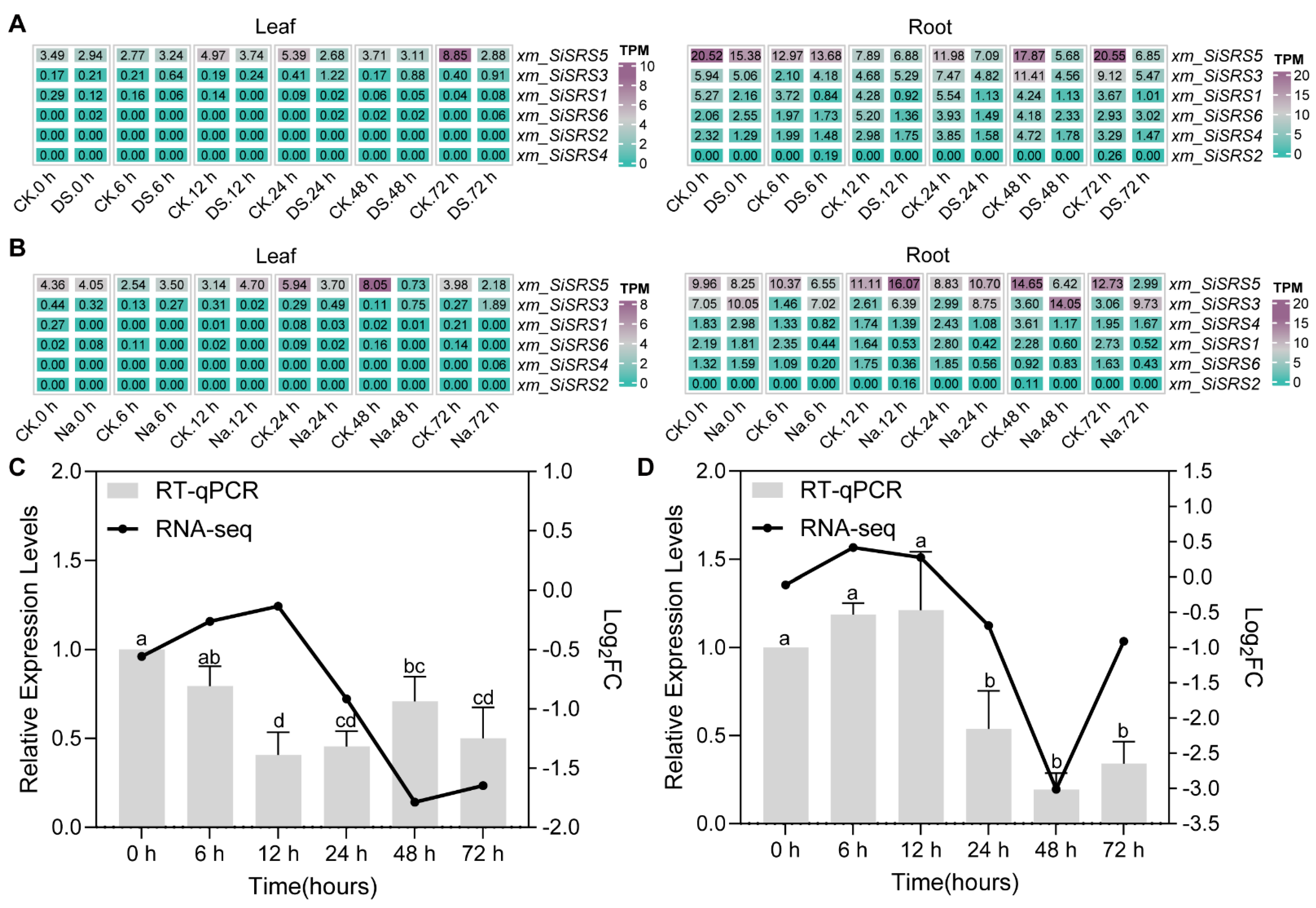
2.6. RNA-seq Data Mining
2.7. RT-qPCR Validation
2.8. Gene Co-Expression and Protein–Protein Interaction Networks
3. Results
3.1. Pan-Genome Distribution of the SiSRS Gene Family
3.2. Pan-Genome-Based Nomenclature for Gene Family Members
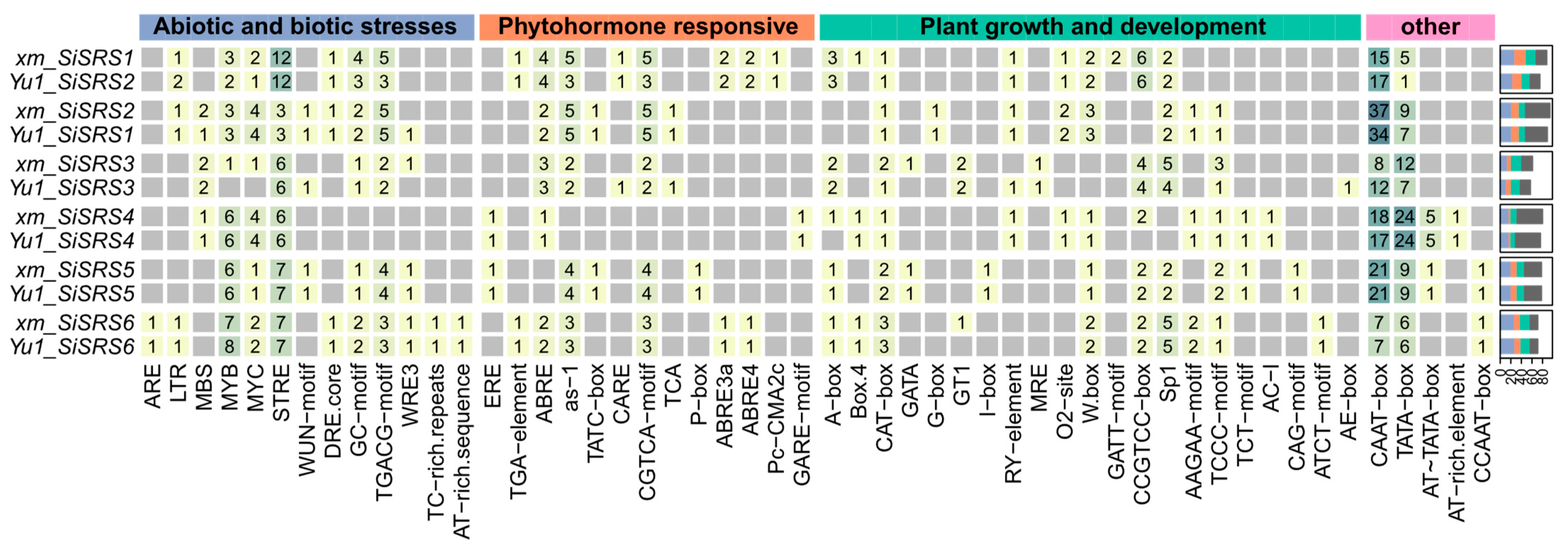
3.3. Pan-Genome-Based Promoter Analysis
3.4. Pan-Genome-Based Motif and Conserved Domain Analysis
3.5. SiSRS Family Members in xm and Yu1
3.6. SiSRS Promoter Architecture and Stress-Responsive Cis-Regulatory Element Profiling in xm and Yu1
3.7. Expression Profiling of xm_SiSRS Genes in Salt and Drought Stress
3.8. Gene Co-Expression Network and PPI Analysis
4. Discussion
4.1. Pan-Genome Analysis as a Paradigm Shift in Gene Family Annotation
4.2. Evolutionary Conservation and Functional Constraints of SiSRS Genes
4.3. xm_SiSRS5 Is the Important SiSRS Member
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fridborg, I.; Kuusk, S.; Moritz, T.; Sundberg, E. The Arabidopsis dwarf mutant shi exhibits reduced gibberellin responses conferred by overexpression of a new putative zinc finger protein. Plant Cell 1999, 11, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Kuusk, S.; Sohlberg, J.J.; Magnus Eklund, D.; Sundberg, E. Functionally redundant SHI family genes regulate Arabidopsis gynoecium development in a dose-dependent manner. Plant J. 2006, 47, 99–111. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Shi, P.; Lv, Y.; Gao, Z.; Chen, G. Gene coexpression network analysis reveals the role of SRS genes in senescence leaf of maize (Zea mays L.). J. Genet. 2020, 99, 3. [Google Scholar] [CrossRef]
- Yang, J.; Xu, P.; Yu, D. Genome-wide identification and characterization of the SHI-related sequence gene family in rice. Evol. Bioinform. 2020, 16, 1176934320941495. [Google Scholar] [CrossRef] [PubMed]
- Youssef, H.M.; Eggert, K.; Koppolu, R.; Alqudah, A.M.; Poursarebani, N.; Fazeli, A.; Sakuma, S.; Tagiri, A.; Rutten, T.; Govind, G.; et al. VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat. Genet. 2017, 49, 157–161. [Google Scholar] [CrossRef]
- Eklund, D.M.; Staldal, V.; Valsecchi, I.; Cierlik, I.; Eriksson, C.; Hiratsu, K.; Ohme-Takagi, M.; Sundstrom, J.F.; Thelander, M.; Ezcurra, I.; et al. The Arabidopsis thaliana STYLISH1 protein acts as a transcriptional activator regulating auxin biosynthesis. Plant Cell 2010, 22, 349–363. [Google Scholar] [CrossRef]
- Jin, J.; He, K.; Tang, X.; Li, Z.; Lv, L.; Zhao, Y.; Luo, J.; Gao, G. An Arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol. Biol. Evol. 2015, 32, 1767–1773. [Google Scholar] [CrossRef]
- Sohlberg, J.J.; Myrenas, M.; Kuusk, S.; Lagercrantz, U.; Kowalczyk, M.; Sandberg, G.; Sundberg, E. STY1 regulates auxin homeostasis and affects apical-basal patterning of the Arabidopsis gynoecium. Plant J. 2006, 47, 112–123. [Google Scholar] [CrossRef]
- Smith, D.L.; Fedoroff, N.V. LRP1, a Gene Expressed in Lateral and Adventitious Root Primordia of Arabidopsis. Plant Cell 1995, 7, 735–745. [Google Scholar]
- Singh, S.; Yadav, S.; Singh, A.; Mahima, M.; Singh, A.; Gautam, V.; Sarkar, A.K. Auxin signaling modulates LATERAL ROOT PRIMORDIUM1 (LRP1) expression during lateral root development in Arabidopsis. Plant J. 2020, 101, 87–100. [Google Scholar] [CrossRef]
- Zhang, Y.; von Behrens, I.; Zimmermann, R.; Ludwig, Y.; Hey, S.; Hochholdinger, F. LATERAL ROOT PRIMORDIA 1 of maize acts as a transcriptional activator in auxin signalling downstream of the Aux/IAA gene rootless with undetectable meristem 1. J. Exp. Bot. 2015, 66, 3855–3863. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.-T.; Xu, H.-H.; Zhang, Q.; Zhang, L.-Y.; Lu, Y.-T. The COP1 Target SHI-RELATED SEQUENCE5 Directly Activates Photomorphogenesis-Promoting Genes. Plant Cell 2018, 30, 2368–2382. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.-T.; Xu, H.-H.; Li, J.; Lu, Y.-T. Auxin abolishes SHI-RELATED SEQUENCE5-mediated inhibition of lateral root development in Arabidopsis. New Phytol. 2020, 225, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Duan, E.; Wang, Y.; Li, X.; Lin, Q.; Zhang, T.; Wang, Y.; Zhou, C.; Zhang, H.; Jiang, L.; Wang, J.; et al. OsSHI1 Regulates Plant Architecture Through Modulating the Transcriptional Activity of IPA1 in Rice. Plant Cell 2019, 31, 1026–1042. [Google Scholar] [CrossRef]
- Zhao, S.P.; Song, X.Y.; Guo, L.L.; Zhang, X.Z.; Zheng, W.J. Genome-Wide Analysis of the Shi-Related Sequence Family and Functional Identification of GmSRS18 Involving in Drought and Salt Stresses in Soybean. Int. J. Mol. Sci. 2020, 21, 1810. [Google Scholar] [CrossRef]
- Sun, C.; Yu, L.; Zhang, S.; Gu, Q.; Wang, M. Genome-wide characterization of the SHORT INTER-NODES/STYLISH and Shi-Related Sequence family in Gossypium hirsutum and functional identification of GhSRS21 under salt stress. Front. Plant Sci. 2022, 13, 1078083. [Google Scholar] [CrossRef]
- Lu, W.; Wang, Y.; Shi, Y.; Liang, Q.; Lu, X.; Su, D.; Xu, X.; Pirrello, J.; Gao, Y.; Huang, B.; et al. Identification of SRS transcription factor family in Solanum lycopersicum, and functional characterization of their responses to hormones and abiotic stresses. BMC Plant Biol. 2023, 23, 495. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Zhang, H.; Liu, Z.; Wang, Y.; Xing, L.; He, Q.; Du, H. Plant pan-genomics: Recent advances, new challenges, and roads ahead. J. Genet. Genom. 2022, 49, 833–846. [Google Scholar] [CrossRef]
- Plissonneau, C.; Hartmann, F.E.; Croll, D. Pangenome analyses of the wheat pathogen Zymoseptoria tritici reveal the structural basis of a highly plastic eukaryotic genome. BMC Biol. 2018, 16, 5. [Google Scholar] [CrossRef]
- Li, X.; Hou, S.; Feng, M.; Xia, R.; Li, J.; Tang, S.; Han, Y.; Gao, J.; Wang, X. MDSi: Multi-omics Database for Setaria italica. BMC Plant Biol. 2023, 23, 223. [Google Scholar] [CrossRef]
- He, Q.; Wang, C.; He, Q.; Zhang, J.; Liang, H.; Lu, Z.; Xie, K.; Tang, S.; Zhou, Y.; Liu, B.; et al. A complete reference genome assembly for foxtail millet and Setaria-db, a comprehensive database for Setaria. Mol. Plant 2024, 17, 219–222. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Tang, S.; Zhi, H.; Chen, J.; Zhang, J.; Liang, H.; Alam, O.; Li, H.; Zhang, H.; Xing, L.; et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet. 2023, 55, 1232–1242. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z. Complex heatmap visualization. Imeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Derbyshire, M.K.; Yamashita, R.A.; Marchler-Bauer, A. NCBI’s conserved domain database and tools for protein domain analysis. Curr. Protoc. Bioinform. 2020, 69, e90. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, 49. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package, Version 1. 2018. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 1 March 2025).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Shang, L.; He, W.; Wang, T.; Yang, Y.; Xu, Q.; Zhao, X.; Yang, L.; Zhang, H.; Li, X.; Lv, Y.; et al. A complete assembly of the rice Nipponbare reference genome. Mol. Plant 2023, 16, 1232–1236. [Google Scholar] [CrossRef]
- Hou, X.; Wang, D.; Cheng, Z.; Wang, Y.; Jiao, Y. A near-complete assembly of an Arabidopsis thaliana genome. Mol. Plant 2022, 15, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cheng, J.; Wu, J.; Chen, J.; Liu, D.; Wang, C.; Ma, S.; Guo, W.; Li, G.; Di, D.; et al. Variation in TaSPL6-D confers salinity tolerance in bread wheat by activating TaHKT1;5-D while preserving yield-related traits. Nat. Genet. 2024, 56, 1257–1269. [Google Scholar] [CrossRef]
- Gui, S.; Wei, W.; Jiang, C.; Luo, J.; Chen, L.; Wu, S.; Li, W.; Wang, Y.; Li, S.; Yang, N.; et al. A pan-Zea genome map for enhancing maize improvement. Genome Biol. 2022, 23, 178. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef]
- Tao, Y.; Luo, H.; Xu, J.; Cruickshank, A.; Zhao, X.; Teng, F.; Hathorn, A.; Wu, X.; Liu, Y.; Shatte, T.; et al. Extensive variation within the pan-genome of cultivated and wild sorghum. Nat. Plants 2021, 7, 766–773. [Google Scholar] [CrossRef]
- Tong, C.; Jia, Y.; Hu, H.; Zeng, Z.; Chapman, B.; Li, C. Pangenome and pantranscriptome as the new reference for gene-family characterization: A case study of basic helix-loop-helix (bHLH) genes in barley. Plant Commun. 2025, 6, 101190. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pak, S.; Yang, J.; Wu, Y.; Li, W.; Feng, H.; Yang, J.; Wei, H.; Li, C. Two high hierarchical regulators, PuMYB40 and PuWRKY75, control the low phosphorus driven adventitious root formation in Populus ussuriensis. Plant Biotechnol. J. 2022, 20, 1561–1577. [Google Scholar] [CrossRef]
- Su, N.; Zhu, A.; Tao, X.; Ding, Z.J.; Chang, S.; Ye, F.; Zhang, Y.; Zhao, C.; Chen, Q.; Wang, J.; et al. Structures and mechanisms of the Arabidopsis auxin transporter PIN3. Nature 2022, 609, 616–621. [Google Scholar] [CrossRef]
- Kim, S.; Koh, J.; Ma, H.; Hu, Y.; Endress, P.K.; Hauser, B.A.; Buzgo, M.; Soltis, P.S.; Soltis, D.E. Sequence and Expression Studies of A-, B-, and E-Class MADS-Box Homologues inEupomatia(Eupomatiaceae): Support for the Bracteate Origin of the Calyptra. Int. J. Plant Sci. 2005, 166, 185–198. [Google Scholar] [CrossRef]
- Jang, S.; Lee, B.; Kim, C.; Kim, S.J.; Yim, J.; Han, J.J.; Lee, S.; Kim, S.R.; An, G. The OsFOR1 gene encodes a polygalacturonase-inhibiting protein (PGIP) that regulates floral organ number in rice. Plant Mol. Biol. 2003, 53, 357–369. [Google Scholar] [CrossRef]
- Rodrigues, O.; Shan, L. Stomata in a state of emergency: H2O2 is the target locked. Trends Plant Sci. 2022, 27, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Schlauch, K.; Tam, R.; Cortes, D.; Torres, M.A.; Shulaev, V.; Dangl, J.L.; Mittler, R. The plant NADPH oxidase RBOHD mediates rapid systemic signaling in response to diverse stimuli. Sci. Signal. 2009, 2, ra45. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, H.; Sun, L.; Jiao, Y.; Zhang, G.; Miao, C.; Hao, F. NADPH oxidase AtrbohD and AtrbohF function in ROS-dependent regulation of Na+/K+ homeostasis in Arabidopsis under salt stress. J. Exp. Bot. 2012, 63, 305–317. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Lei, C.; Zhang, Q.; Guo, X.; Cui, X.; Wang, X.; Li, X.; Gao, J. Pan-Genome-Based Characterization of the SRS Transcription Factor Family in Foxtail Millet. Plants 2025, 14, 1257. https://doi.org/10.3390/plants14081257
Li R, Lei C, Zhang Q, Guo X, Cui X, Wang X, Li X, Gao J. Pan-Genome-Based Characterization of the SRS Transcription Factor Family in Foxtail Millet. Plants. 2025; 14(8):1257. https://doi.org/10.3390/plants14081257
Chicago/Turabian StyleLi, Ruimiao, Cuiyun Lei, Qiang Zhang, Xiaomeng Guo, Xiting Cui, Xingchun Wang, Xukai Li, and Jianhua Gao. 2025. "Pan-Genome-Based Characterization of the SRS Transcription Factor Family in Foxtail Millet" Plants 14, no. 8: 1257. https://doi.org/10.3390/plants14081257
APA StyleLi, R., Lei, C., Zhang, Q., Guo, X., Cui, X., Wang, X., Li, X., & Gao, J. (2025). Pan-Genome-Based Characterization of the SRS Transcription Factor Family in Foxtail Millet. Plants, 14(8), 1257. https://doi.org/10.3390/plants14081257