


Aerospace Mutagenized Tea Tree Increases Rhizospheric Microorganisms, Enhances Nutrient Conversion Capacity and Promotes Growth

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
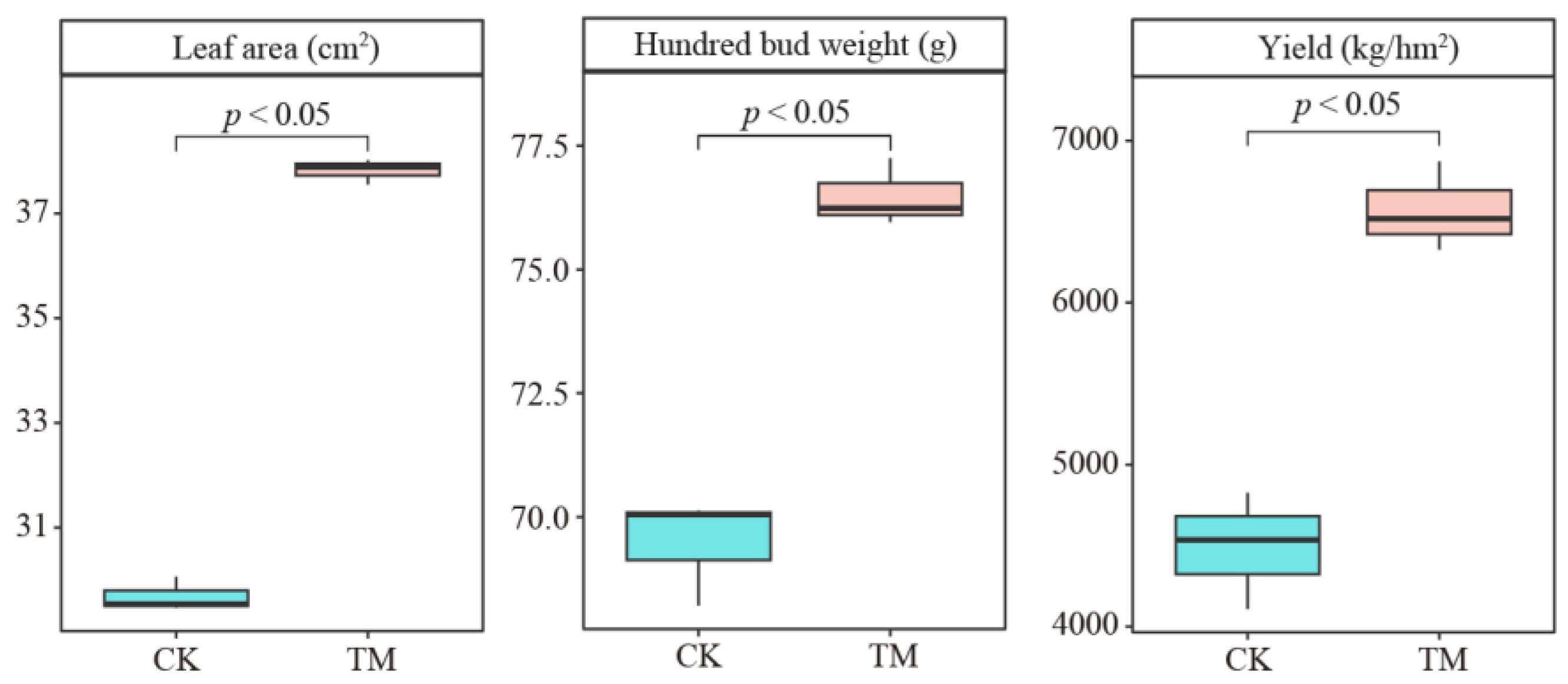
2.1. Effect of Aerospace Mutagenesis on the Growth of Tea Trees
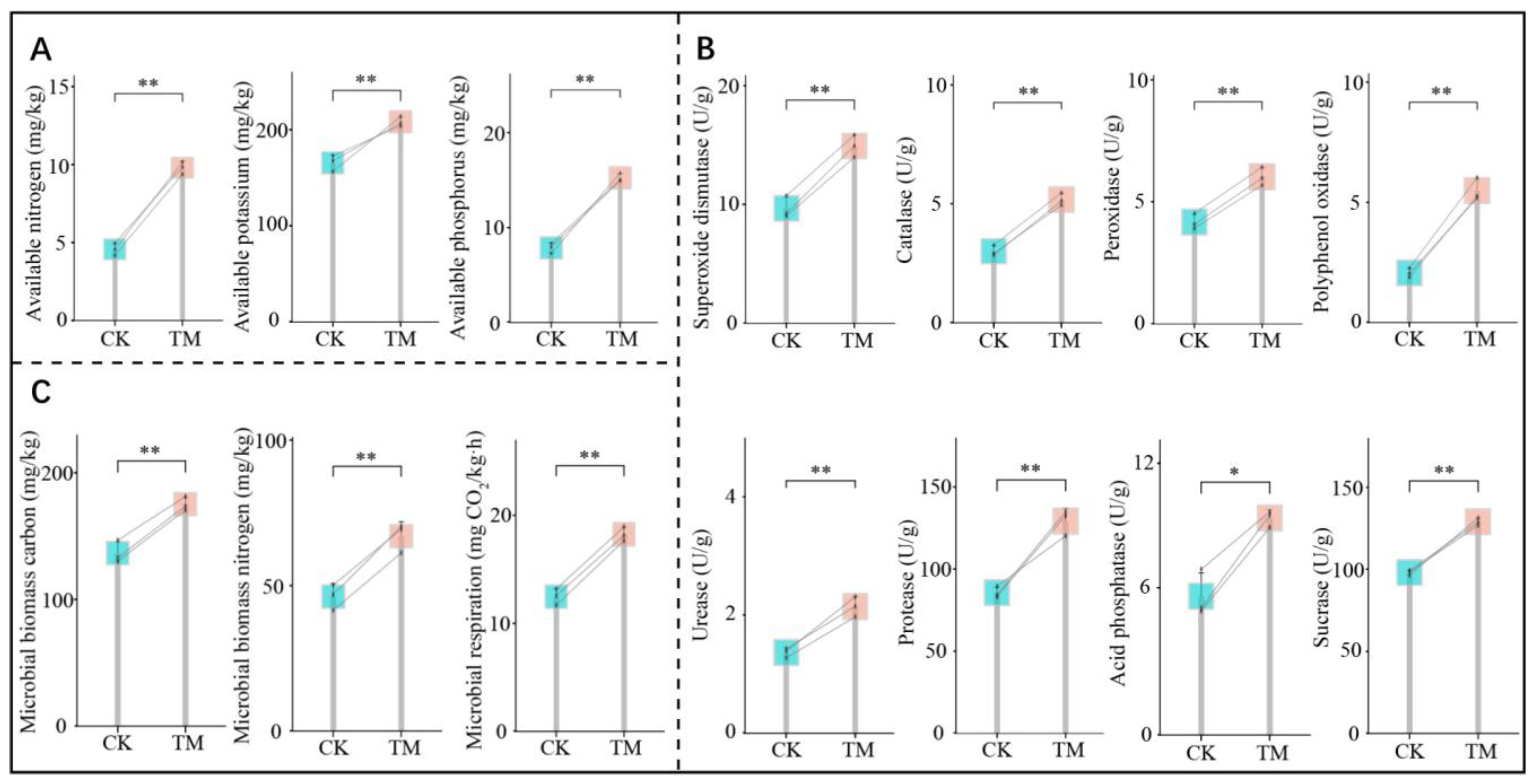
2.2. Effects of Aerospace Mutagenesis on Nutrient Contents, Enzyme Activities, and Microbial Physiological Indices of the Rhizosphere Soil of Tea Trees
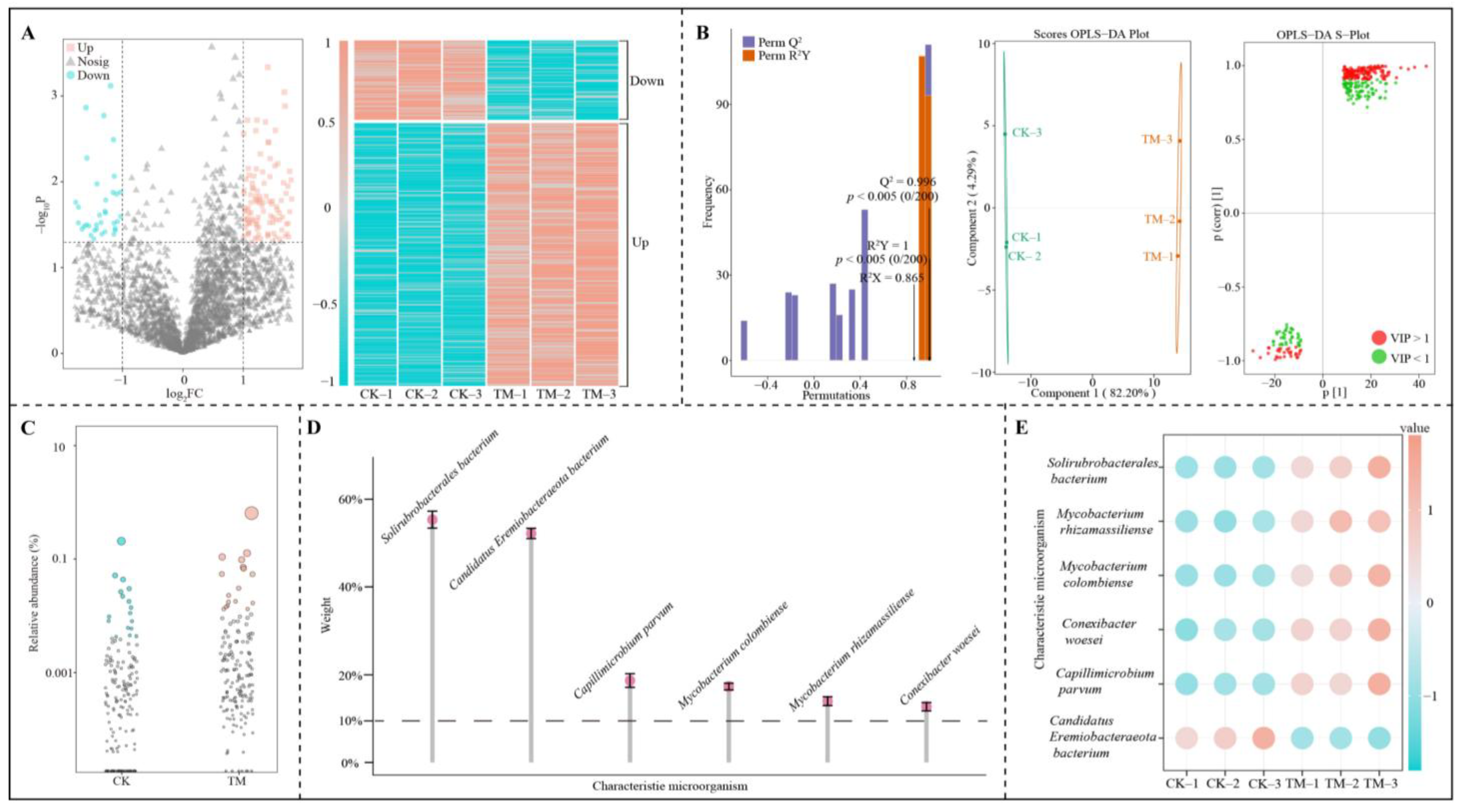
2.3. Soil Microbial Macro-Genome Sequencing Analysis
2.4. Effect of Aerospace Mutagenesis on the Microbial Diversity of Rhizosphere Soil of Tea Trees
2.5. Modular Network Node Analysis of Microorganisms in the Rhizosphere Soil of Tea Tree
2.6. Screening of Characteristic Microorganisms in the Rhizosphere Soil of Tea Trees After Aerospace Mutagenesis
2.7. Functional Prediction and Intensity Analysis of Characteristic Microorganisms
2.8. Interaction Analysis of Characteristic Microorganisms and Their Functions with Different Indices
3. Materials and Methods
3.1. Experimental Site and Sample Sampling
3.2. Determination of Growth Indices of Tea Trees
3.3. Determination of Soil Available Nutrient Content
3.4. Determination of Soil Enzyme Activity
3.5. Determination of Physiological Indices of Soil Microorganisms
3.6. Macrogenome Sequencing and Bioinformatics Analysis of Soil Microorganisms
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Udage, A.C. Introduction to plant mutation breeding: Different approaches and mutagenic agents. J. Agric. Sci. 2021, 16, 466–483. [Google Scholar] [CrossRef]
- Lamichhane, S.; Thapa, S. Advances from conventional to modern plant breeding methodologies. Plant Breed. Biotechnol. 2022, 10, 1–14. [Google Scholar] [CrossRef]
- Prasad, B.; Richter, P.; Vadakedath, N.; Mancinelli, R.; Krüger, M.; Strauch, S.M.; Grimm, D.; Darriet, P.; Chapel, J.P.; Cohen, J.; et al. Exploration of space to achieve scientific breakthroughs. Biotechnol. Adv. 2020, 43, 107572. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Richter, P.; Vadakedath, N.; Haag, F.W.; Strauch, S.M.; Mancinelli, R.; Schwarzwälder, A.; Etcheparre, E.; Gaume, N.; Lebert, M. How the space environment influences organisms: An astrobiological perspective and review. Int. J. Astrobiol. 2021, 20, 159–177. [Google Scholar] [CrossRef]
- Furukawa, S.; Nagamatsu, A.; Nenoi, M.; Fujimori, A.; Kakinuma, S.; Katsube, T.; Wang, B.; Tsuruoka, C.; Shirai, T.; Nakamura, A.; et al. Space radiation biology for “Living in Space”. BioMed Res. Int. 2020, 2020, 4703286. [Google Scholar] [CrossRef]
- Li, J.; Liu, M.; Wang, P.; Zhang, J.; Xue, H.; Guo, Y. Effects of space conditions on mutation and inheritance of tomato. Space Med. Med. Eng. 2000, 2, 114–118. [Google Scholar] [CrossRef]
- Ma, L.; Kong, F.; Sun, K.; Wang, T.; Guo, T. From classical radiation to modern radiation: Past, present, and future of radiation mutation breeding. Front. Public Health 2021, 9, 768071. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.; Wu, L.; Chen, R.; Luo, S. The complete chloroplast genome sequence of Camellia sinensis cv. Dahongpao: A most famous variety of Wuyi tea (Synonym: Thea bohea L.). Mitochondrial DNA Part B 2021, 6, 3–5. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Wang, W.; Wang, H.; Cai, J.; Wang, M.; Li, D.; Zhang, B.; Huang, K. Effects of space treatment on biological and growth characteristics of Camellia sinensis. Chin. Bull. Bot. 2020, 55, 564–572. [Google Scholar] [CrossRef]
- Jia, M.; Chen, Y.; Zhang, Q.; Wang, Y.; Li, M.; Pang, X.; Hong, L.; Lin, S.; Jia, X.; Ye, J.; et al. Changes in the growth and physiological property of tea tree after aviation mutagenesis and screening and functional verification of its characteristic hormones. Front. Plant Sci. 2024, 15, 1402451. [Google Scholar] [CrossRef]
- Oldroyd, G.E.; Leyser, O. A plant’s diet, surviving in a variable nutrient environment. Science 2020, 368, eaba0196. [Google Scholar] [CrossRef] [PubMed]
- Cochavi, A.; Cohen, I.H.; Rachmilevitch, S. The role of different root orders in nutrient uptake. Environ. Exp. Bot. 2020, 179, 104212. [Google Scholar] [CrossRef]
- Briat, J.F.; Gojon, A.; Plassard, C.; Rouached, H.; Lemaire, G. Reappraisal of the central role of soil nutrient availability in nutrient management in light of recent advances in plant nutrition at crop and molecular levels. Eur. J. Agron. 2020, 116, 126069. [Google Scholar] [CrossRef]
- Singh, S.K.; Wu, X.; Shao, C.; Zhang, H.M. Microbial enhancement of plant nutrient acquisition. Stress Biol. 2022, 2, 3. [Google Scholar] [CrossRef]
- Prommer, J.; Walker, T.W.; Wanek, W.; Braun, J.; Zezula, D.; Hu, Y.; Hofhansl, F.; Richter, A. Increased microbial growth, biomass, and turnover drive soil organic carbon accumulation at higher plant diversity. Glob. Change Biol. 2020, 26, 669–681. [Google Scholar] [CrossRef]
- Philippot, L.; Chenu, C.; Kappler, A.; Rillig, M.C.; Fierer, N. The interplay between microbial communities and soil properties. Nat. Rev. Microbiol. 2024, 22, 226–239. [Google Scholar] [CrossRef]
- Yadav, A.N.; Kour, D.; Kaur, T.; Devi, R.; Yadav, A.; Dikilitas, M.; Abdel-Azeem, A.; Ahluwalia, A.; Saxena, A.K. Biodiversity, and biotechnological contribution of beneficial soil microbiomes for nutrient cycling, plant growth improvement and nutrient uptake. Biocatal. Agric. Biotechnol. 2021, 33, 102009. [Google Scholar] [CrossRef]
- Mitra, D.; Mondal, R.; Khoshru, B.; Senapati, A.; Radha, T.K.; Mahakur, B.; Uniyal, N.; Myo, E.; Boutaj, H.; Guerra, B.; et al. Actinobacteria-enhanced plant growth, nutrient acquisition, and crop protection: Advances in soil, plant, and microbial multifactorial interactions. Pedosphere 2022, 32, 149–170. [Google Scholar] [CrossRef]
- Dotaniya, M.L.; Meena, V.D. Rhizosphere effect on nutrient availability in soil and its uptake by plants: A review. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2015, 85, 1–12. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Wang, L.; Bai, H.; Wu, B.; Hao, Y.; Zhang, W. Research on the effects of space Mutagenesis on yield and quality of sorghum. J. Agric. 2015, 5, 19–22. [Google Scholar] [CrossRef]
- Soares, J.C.; Santos, C.S.; Carvalho, S.M.; Pintado, M.M.; Vasconcelos, M.W. Preserving the nutritional quality of crop plants under a changing climate: Importance and strategies. Plant Soil 2019, 443, 1–26. [Google Scholar] [CrossRef]
- Zielewicz, W.; Swędrzyński, A.; Dobrzyński, J.; Swędrzyńska, D.; Kulkova, I.; Wierzchowski, P.S.; Wróbel, B. Effect of forage plant mixture and biostimulants application on the yield, changes of botanical composition, and microbiological soil activity. Agronomy 2021, 11, 1786. [Google Scholar] [CrossRef]
- Tian, J.; Ge, F.; Zhang, D.; Deng, S.; Liu, X. Roles of phosphate solubilizing microorganisms from managing soil phosphorus deficiency to mediating biogeochemical P cycle. Biology 2021, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Dong, H.; Wang, Z.; Tao, Y. Effect of straw returning and nitrogen application rate on soil enzymatic activities. Agric. Res. 2023, 12, 163–171. [Google Scholar] [CrossRef]
- Lin, S.; Liu, Z.; Wang, Y.; Li, J.; Wang, G.; Ye, J.; Wang, H.; He, H. Soil metagenomic analysis on changes of functional genes and microorganisms involved in nitrogen-cycle processes of acidified tea soils. Front. Plant Sci. 2022, 13, 998178. [Google Scholar] [CrossRef]
- Macedo, G.; van Veelen, H.P.J.; Hernandez-Leal, L.; van der Maas, P.; Heederik, D.; Mevius, D.; Bossers, A.; Schmitt, H. Targeted metagenomics reveals inferior resilience of farm soil resistome compared to soil microbiome after manure application. Sci. Total Environ. 2021, 770, 145399. [Google Scholar] [CrossRef]
- Conrad, R.E.; Viver, T.; Gago, J.F.; Hatt, J.K.; Venter, S.N.; Rossello-Mora, R.; Konstantinidis, K.T. Toward quantifying the adaptive role of bacterial pangenomes during environmental perturbations. ISME J. 2022, 16, 1222–1234. [Google Scholar] [CrossRef]
- Yuan, M.M.; Guo, X.; Wu, L.; Zhang, Y.A.; Xiao, N.; Ning, D.; Shi, Z.; Zhou, X.; Wu, L.; Yang, Y.; et al. Climate warming enhances microbial network complexity and stability. Nat. Clim. Change 2021, 11, 343–348. [Google Scholar] [CrossRef]
- Willis, A.D. Rarefaction, alpha diversity, and statistics. Front. Microbiol. 2019, 10, 2407. [Google Scholar] [CrossRef]
- Xiao, X.; Zhang, N.; Ni, H.; Yang, Y.; Zhou, J.; Sun, B.; Liang, Y. A latitudinal gradient of microbial β-diversity in continental paddy soils. Glob. Ecol. Biogeogr. 2021, 30, 909–919. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, K.; Li, K.; Jin, Y.; He, X. Deciphering the diversity patterns and community assembly of rare and abundant bacterial communities in a wetland system. Sci. Total Environ. 2022, 838, 156334. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Li, D.; Ding, J.; Xiao, X.; Liang, Y. Microbial coexistence in the rhizosphere and the promotion of plant stress resistance: A review. Environ. Res. 2023, 222, 115298. [Google Scholar] [CrossRef] [PubMed]
- Siles, J.A.; García-Sánchez, M.; Gómez-Brandón, M. Studying microbial communities through co-occurrence network analyses during processes of waste treatment and in organically amended soils: A review. Microorganisms 2021, 9, 1165. [Google Scholar] [CrossRef] [PubMed]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef]
- Xun, W.; Liu, Y.; Li, W.; Ren, Y.; Xiong, W.; Xu, Z.; Zhang, N.; Miao, Y.; Shen, Q.; Zhang, R. Specialized metabolic functions of keystone taxa sustain soil microbiome stability. Microbiome 2021, 9, 35. [Google Scholar] [CrossRef]
- Jia, M.; Cheng, P.; Wang, Y.; Pang, X.; Li, M.; Hong, L.; Zhang, Q.; Chen, Y.; Jia, X.; Ye, J.; et al. Effects of aviation mutagenesis on soil chemical indexes, enzyme activities, and metabolites of Dahongpao (Camellia sinensis) tea trees. Plants 2024, 13, 1291. [Google Scholar] [CrossRef]
- Frantzeskakis, L.; Di Pietro, A.; Rep, M.; Schirawski, J.; Wu, C.H.; Panstruga, R. Rapid evolution in plant–microbe interactions–a molecular genomics perspective. New Phytol. 2020, 225, 1134–1142. [Google Scholar] [CrossRef]
- Zhu, G.; Schmidt, O.; Luan, L.; Xue, J.; Fan, J.; Geisen, S.; Sun, B.; Jiang, Y. Bacterial keystone taxa regulate carbon metabolism in the earthworm gut. Microbiol. Spectr. 2022, 10, e01081-22. [Google Scholar] [CrossRef]
- Huang, J.N.; Xu, L.; Wen, B.; Gao, J.Z.; Chen, Z.Z. Reshaping the plastisphere upon deposition: Promote N2O production through affecting sediment microbial communities in aquaculture pond. J. Hazard. Mater. 2024, 465, 133290. [Google Scholar] [CrossRef]
- Pavlik, I.; Ulmann, V.; Hubelova, D.; Weston, R.T. Nontuberculous mycobacteria as sapronoses: A review. Microorganisms 2022, 10, 1345. [Google Scholar] [CrossRef]
- Torvinen, E.; Lehtola, M.J.; Martikainen, P.J.; Miettinen, I.T. Survival of Mycobacterium avium in drinking water biofilms as affected by water flow velocity, availability of phosphorus, and temperature. Appl. Environ. Microbiol. 2007, 73, 6201–6207. [Google Scholar] [CrossRef] [PubMed]
- Jien, S.H.; Kuo, Y.L.; Liao, C.S.; Wu, Y.T.; Igalavithana, A.D.; Tsang, D.C.; Ok, Y.S. Effects of field scale in situ biochar incorporation on soil environment in a tropical highly weathered soil. Environ. Pollut. 2021, 272, 116009. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Yang, J.; Su, S.; Liu, Y.; Feng, J.; Sheng, Y.; Wang, T.; Pan, J.; Tang, L.; Chen, L.; et al. Distribution of genes and microbial taxa related to soil phosphorus cycling across soil depths in subtropical forests. Forests 2023, 14, 1665. [Google Scholar] [CrossRef]
- Dal Bello, M.; Lee, H.; Goyal, A.; Gore, J. Resource–diversity relationships in bacterial communities reflect the network structure of microbial metabolism. Nat. Ecol. Evol. 2021, 5, 1424–1434. [Google Scholar] [CrossRef]
- Mokrani, S.; Nabti, E.H. Recent status in production, biotechnological applications, commercial aspects, and future prospects of microbial enzymes: A comprehensive review. Int. J. Agric. Food Sci. 2024, 10, 6–20. [Google Scholar] [CrossRef]
- Nunan, N.; Schmidt, H.; Raynaud, X. The ecology of heterogeneity: Soil bacterial communities and C dynamics. Philos. Trans. R. Soc. B 2020, 375, 20190249. [Google Scholar] [CrossRef]
- Qin, S.; Gao, W.; Jing, Y.; Quan, W.; Cai, K. Soil pseudotargeted metabolomics reveals that planting years of masson pine (Pinus massoniana) affect soil metabolite profiles and metabolic pathways. Plant Soil 2024, 496, 505–520. [Google Scholar] [CrossRef]
- Wang, C.; Xue, L.; Dong, Y.; Jiao, R. Soil organic carbon fractions, C-cycling hydrolytic enzymes, and microbial carbon metabolism in Chinese fir plantations. Sci. Total Environ. 2021, 758, 143695. [Google Scholar] [CrossRef]
- Sible, C.N.; Seebauer, J.R.; Below, F.E. Plant biostimulants: A categorical review, their implications for row crop production, and relation to soil health indicators. Agronomy 2021, 11, 1297. [Google Scholar] [CrossRef]
- Bi, B.; Wang, K.; Zhang, H.; Wang, Y.; Fei, H.; Pan, R.; Han, F. Plants use rhizosphere metabolites to regulate soil microbial diversity. Land Degrad. Dev. 2021, 32, 5267–5280. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Z.; Hu, W.; Bai, H.; Ma, L.; Lv, X.; Zhou, Z.; Meng, Y. Crop residue return improved soil nitrogen availability by increasing amino acid and mineralization under appropriate N fertilization. Land Degrad. Dev. 2022, 33, 2197–2207. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.; Li, J.; Lin, S.; Jia, X.; Zhang, Q.; Ye, J.; Wang, H.; Wu, Z. Study on the effect of pH on rhizosphere soil fertility and the aroma quality of tea trees and their interactions. Agriculture 2023, 13, 1739. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Q.; Shi, L.; Wang, Y.; Li, M.; Chen, Y.; Zhang, M.; Chen, J.; Chen, M.; Jia, X.; et al. Joint analysis of transcriptome and hormone metabolome on the mechanism of pruning effect on tea tree (Camellia sinensis) growth. Ind. Crops Prod. 2024, 218, 118929. [Google Scholar] [CrossRef]
- Jia, X.; Lin, S.; Zhang, Q.; Wang, Y.; Hong, L.; Li, M.; Zhang, S.; Wang, T.; Jia, M.; Luo, Y.; et al. The ability of different tea tree germplasm resources in south China to aggregate rhizosphere soil characteristic fungi affects tea quality. Plants 2024, 13, 2029. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, Z.; Peng, S.; Yuan, Y.; Wang, X. Influences of lithium on soil properties and enzyme activities. Chemosphere 2023, 313, 137458. [Google Scholar] [CrossRef]
- Schnecker, J.; Baldaszti, L.; Gündler, P.; Pleitner, M.; Sandén, T.; Simon, E.; Spiegel, F.; Spiegel, H.; Malo, C.U.; Boltenstern, S.Z.; et al. Seasonal dynamics of soil microbial growth, respiration, biomass, and carbon use efficiency in temperate soils. Geoderma 2023, 440, 116693. [Google Scholar] [CrossRef]
- Wilton, R.; Szalay, A.S. Performance optimization in DNA short-read alignment. Bioinformatics 2022, 38, 2081–2087. [Google Scholar] [CrossRef]
- Goussarov, G.; Mysara, M.; Vandamme, P.; Van Houdt, R. Introduction to the principles and methods underlying the recovery of metagenome-assembled genomes from metagenomic data. Microbiologyopen 2022, 11, e1298. [Google Scholar] [CrossRef]
- Ji, Y.; Xi, H.; Zhao, Z.; Jiang, Q.; Chen, C.; Wang, X.; Li, F.; Li, N.; Sun, C.; Feng, X.; et al. Metagenomics analysis reveals potential pathways and drivers of piglet gut phage-mediated transfer of ARGs. Sci. Total Environ. 2023, 859, 160304. [Google Scholar] [CrossRef]
- Yones, C.; Raad, J.; Bugnon, L.A.; Milone, D.H.; Stegmayer, G. High precision in microRNA prediction: A novel genome-wide approach with convolutional deep residual networks. Comput. Biol. Med. 2021, 134, 104448. [Google Scholar] [CrossRef]
- Kondratenko, Y.; Korobeynikov, A.; Lapidus, A. CDSnake: Snakemake pipeline for retrieval of annotated OTUs from paired-end reads using CD-HIT utilities. BMC Bioinform. 2020, 21, 303. [Google Scholar] [CrossRef]
- Sun, S.; Jones, R.B.; Fodor, A.A. Inference-based accuracy of metagenome prediction tools varies across sample types and functional categories. Microbiome 2020, 8, 46. [Google Scholar] [CrossRef]
- Xu, R.; Rajeev, S.; Salvador, L.C. The selection of software and database for metagenomics sequence analysis impacts the outcome of microbial profiling and pathogen detection. PLoS ONE 2023, 18, e0284031. [Google Scholar] [CrossRef]
- Portik, D.M.; Brown, C.T.; Pierce-Ward, N.T. Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets. BMC Bioinform. 2022, 23, 541. [Google Scholar] [CrossRef]
- Liu, G.; Wu, C.; Abrams, W.R.; Li, Y. Structural and functional characteristics of the microbiome in deep-dentin caries. J. Dent. Res. 2020, 99, 713–720. [Google Scholar] [CrossRef]
- Wickham, H. Advanced R, 2nd ed.; Chapman and Hall/CRC The R Series; Taylor and Francis: Abingdon, UK, 2019. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.; Wang, Y.; Wang, Y.; Hong, L.; Qiu, M.; Luo, Y.; Zhang, Q.; Wang, T.; Jia, X.; Wang, H.; et al. Aerospace Mutagenized Tea Tree Increases Rhizospheric Microorganisms, Enhances Nutrient Conversion Capacity and Promotes Growth. Plants 2025, 14, 981. https://doi.org/10.3390/plants14070981
Cheng W, Wang Y, Wang Y, Hong L, Qiu M, Luo Y, Zhang Q, Wang T, Jia X, Wang H, et al. Aerospace Mutagenized Tea Tree Increases Rhizospheric Microorganisms, Enhances Nutrient Conversion Capacity and Promotes Growth. Plants. 2025; 14(7):981. https://doi.org/10.3390/plants14070981
Chicago/Turabian StyleCheng, Weiting, Yulin Wang, Yuhua Wang, Lei Hong, Miaoen Qiu, Yangxin Luo, Qi Zhang, Tingting Wang, Xiaoli Jia, Haibin Wang, and et al. 2025. "Aerospace Mutagenized Tea Tree Increases Rhizospheric Microorganisms, Enhances Nutrient Conversion Capacity and Promotes Growth" Plants 14, no. 7: 981. https://doi.org/10.3390/plants14070981
APA StyleCheng, W., Wang, Y., Wang, Y., Hong, L., Qiu, M., Luo, Y., Zhang, Q., Wang, T., Jia, X., Wang, H., & Ye, J. (2025). Aerospace Mutagenized Tea Tree Increases Rhizospheric Microorganisms, Enhances Nutrient Conversion Capacity and Promotes Growth. Plants, 14(7), 981. https://doi.org/10.3390/plants14070981