
Characterizing Wheat Rhizosphere Bacterial Microbiome Dynamics Under Salinity Stress: Insights from 16S rRNA Metagenomics for Enhancing Stress Tolerance

 , ,
, , 
Abstract
1. Introduction
2. Material and Methods
2.1. Microbial Community DNA Extraction
2.2. Metagenomics Amplicon Sequencing
2.2.1. 16S rRNA Gene Amplification
2.2.2. Library Preparation and Next-Generation Sequencing
2.3. Sequence Analysis
Data Filtering and Normalization
2.4. Visual Exploration Profiling
2.5. Community Profiling
Alpha, Beta Diversity, and Core Microbiome
2.6. Interactions Through Spearman Rank Correlation Analysis
2.7. Differential Abundance Analysis
3. Results
3.1. Microbial Abundance Data
3.2. Visual Exploration
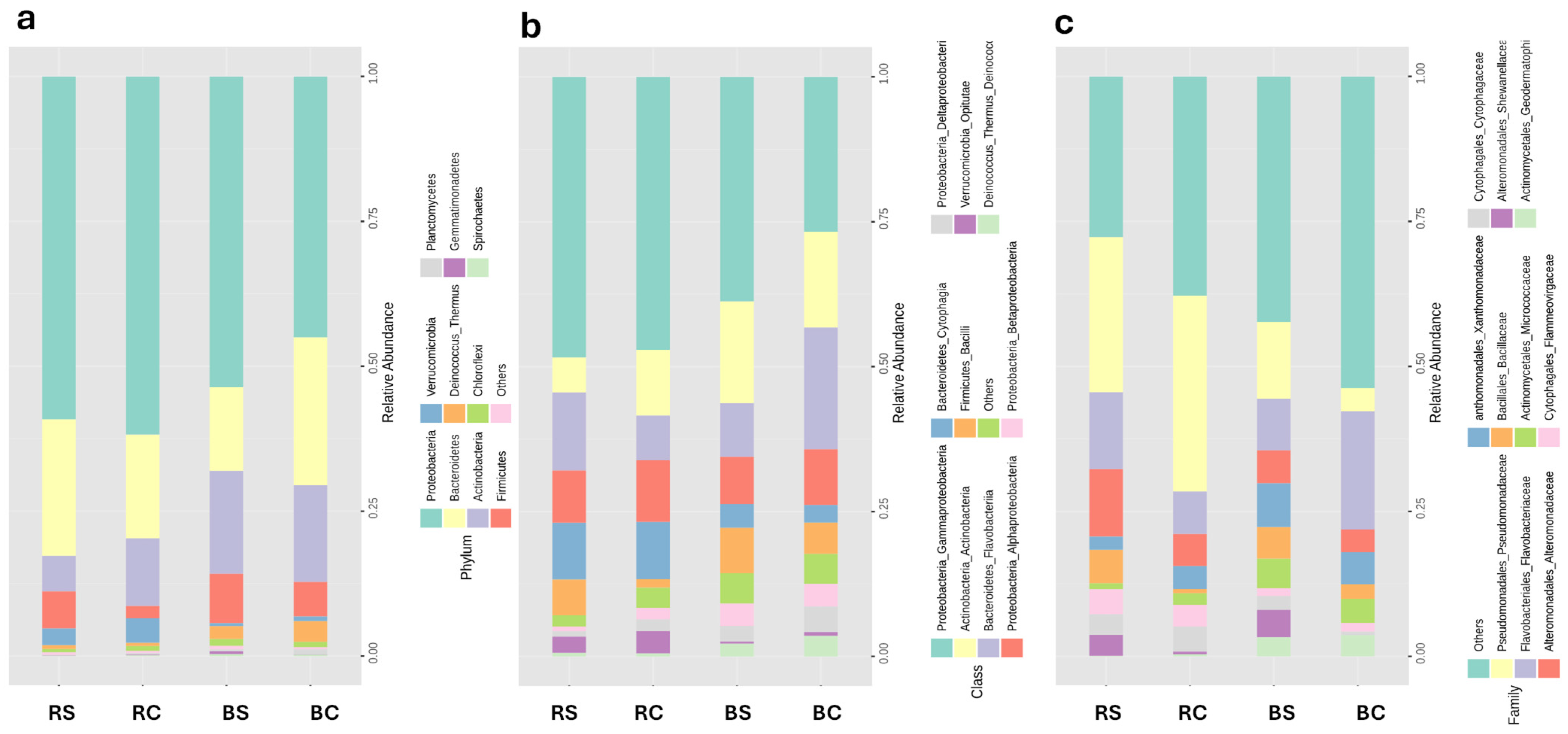
3.3. Microbial Community Profiling
3.3.1. Alpha Diversity Assessment
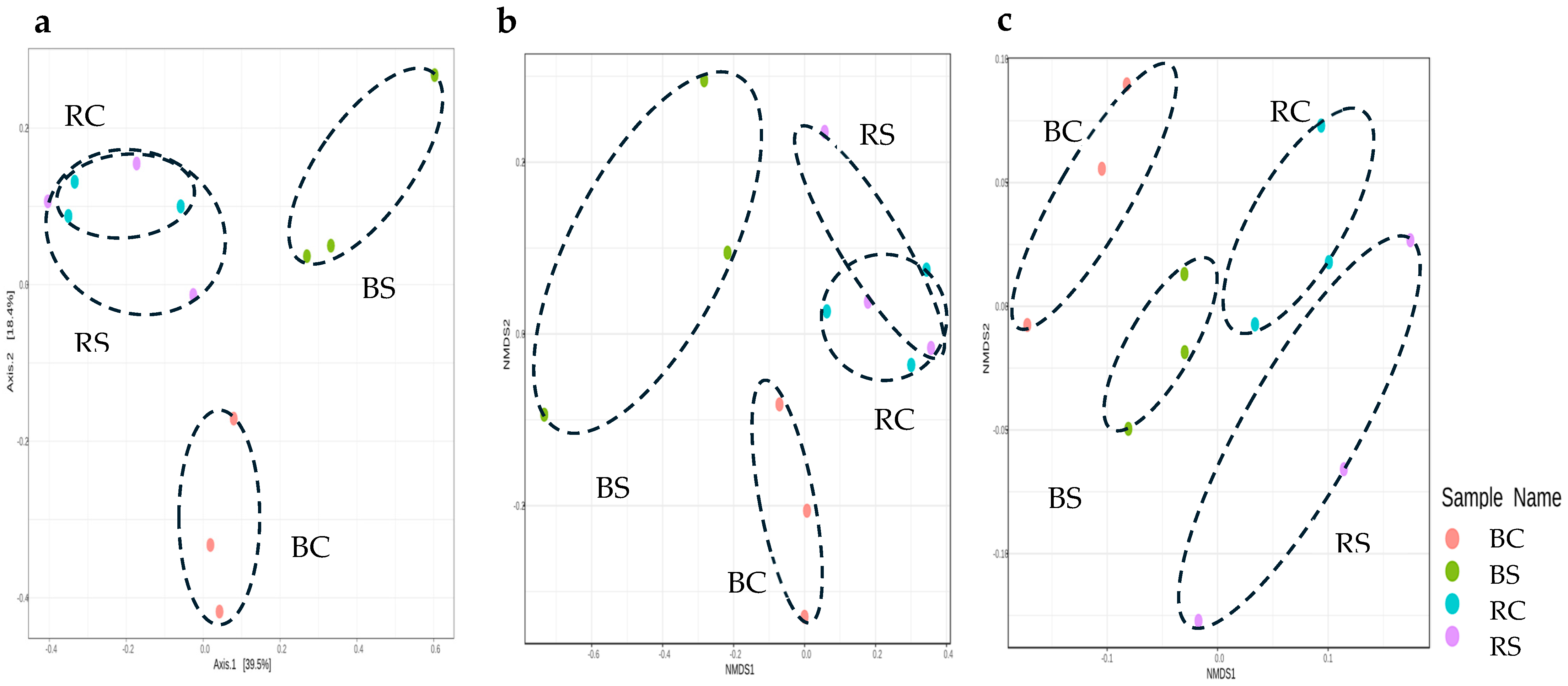
3.3.2. Beta Diversity Assessment
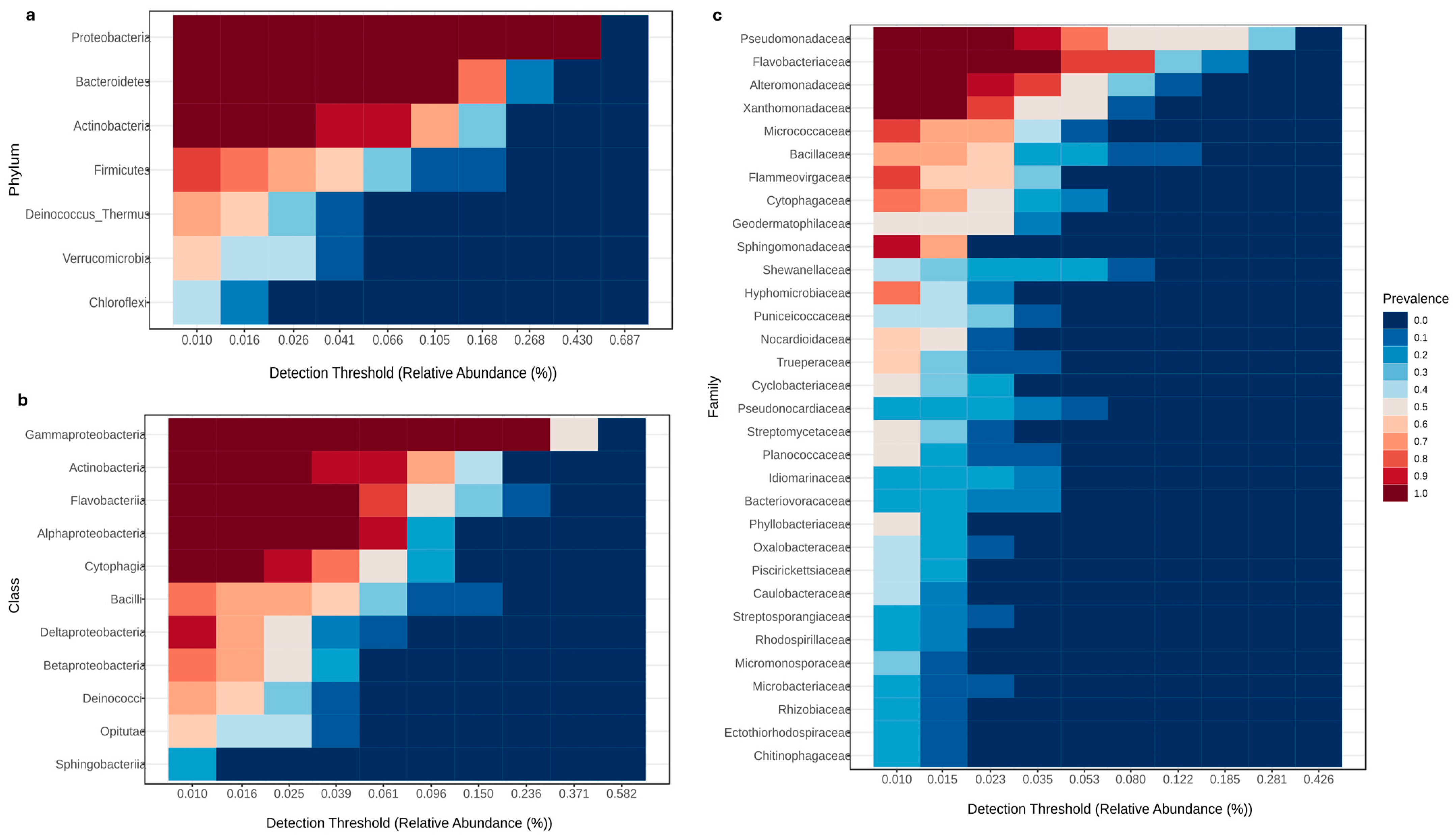
3.3.3. Core Microbiome Analysis
3.4. Correlation Analysis
3.5. Differential Abundance Profiling (DAP)
- a.
- Microbial recruiting:
- 1
- Wheat microbial recruitment under normal conditions: BC vs. RC:
- 2
- Wheat microbial recruitment under saline conditions: BS vs. RS:
- b.
- Salinity-Induced Microbial Shifts:
- c.
- Microbial signature:
4. Discussion
4.1. Assessment of Abundance Count Data
4.2. Alpha and Beta Diversity Analysis
4.3. Consistent Core Microbiome Communities
4.4. Stability of Microbial Communities
4.5. Variation in Bacterial Abundance Across Treatments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Manhou, K.; Moussadek, R.; Yachou, H.; Zouahri, A.; Douaik, A.; Hilal, I.; Ghanimi, A.; Hmouni, D.; Dakak, H. Assessing the Impact of Saline Irrigation Water on Durum Wheat (cv. Faraj) Grown on Sandy and Clay Soils. Agronomy 2024, 14, 2865. [Google Scholar] [CrossRef]
- Nazir, M. Salinity stress resistance in wheat. World J. Biol. Biotechnol. 2021, 6, 27–31. [Google Scholar]
- Tadesse, W.; Amri, A.; Ogbonnaya, F.C.; Sanchez-Garcia, M.; Sohail, Q.; Baum, M. Wheat. In Genetic and Genomic Resources for Grain Cereals Improvement; Elsevier: Amsterdam, The Netherlands, 2016; pp. 81–124. [Google Scholar]
- Tadesse, W.; Halila, H.; Jamal, M.; Hanafi, S.; Assefa, S.; Oweis, T.; Baum, M. Role of sustainable wheat production to ensure food security in the CWANA region. J. Exp. Biol. Agric. Sci. 2017, 5, 15–32. [Google Scholar]
- Seleiman, M.F.; Aslam, M.T.; Alhammad, B.A.; Hassan, M.U.; Maqbool, R.; Chattha, M.U.; Khan, I.; Gitari, H.I.; Uslu, O.S.; Roy, R.; et al. Salinity stress in wheat: Effects, mechanisms and management strategies. Phyton-Int. J. Exp. Bot. 2022, 91, 667–694. [Google Scholar] [CrossRef]
- Sharma, P.; Pandey, R.; Chauhan, N.S. Unveiling wheat growth promotion potential of phosphate solubilizing Pantoea agglomerans PS1 and PS2 through genomic, physiological, and metagenomic characterizations. Front. Microbiol. 2024, 15, 1467082. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hu, L.; Rozhkova, T.; Li, C. Enhancing resistance to salinity in wheat by using Streptomyces sp. HU2014. Agronomy 2023, 14, 39. [Google Scholar] [CrossRef]
- Gao, Y.; Zou, H.; Wang, B.; Yuan, F. Progress and applications of plant growth-promoting bacteria in salt tolerance of crops. Int. J. Mol. Sci. 2022, 23, 7036. [Google Scholar] [CrossRef]
- Zahra, S.T.; Tariq, M.; Abdullah, M.; Azeem, F.; Ashraf, M.A. Dominance of Bacillus species in the wheat (Triticum aestivum L.) rhizosphere and their plant growth promoting potential under salt stress conditions. PeerJ 2023, 11, e14621. [Google Scholar]
- Xu, F.; Liang, Y.; Wang, X.; Guo, Y.; Tang, K.; Feng, F. Synergic mitigation of saline-alkaline stress in wheat plant by silicon and Enterobacter sp. FN0603. Front. Microbiol. 2023, 13, 1100232. [Google Scholar]
- Peng, W.; Dong, Y.; Wang, J.; Wang, S.; Wang, N. Effects of exogenous solution treatment on germination, antioxidation and flavonoid biosynthesis of Tartary buckwheat (Fagopyrum tataricum (L.) Gaertn.). Food Biosci. 2023, 56, 103367. [Google Scholar]
- Liu, H.; Khan, M.Y.; Carvalhais, L.C.; Delgado-Baquerizo, M.; Yan, L.; Crawford, M.; Dennis, P.G.; Singh, B.; Schenk, P.M. Soil amendments with ethylene precursor alleviate negative impacts of salinity on soil microbial properties and productivity. Sci. Rep. 2019, 9, 6892. [Google Scholar]
- Shi, S.; Ge, M.; Xiong, Y.; Zhang, Y.; Li, W.; Wang, J.; He, E.; Liu, Z.; Zhou, D. Characteristics and Functions of Rhizosphere Bacterial Communities of C. sinensis Cv. Shifocui Tea Tree With Cold Resistance Characteristics in Anhui Province, China. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Singh, A.; Mazahar, S.; Chapadgaonkar, S.S.; Giri, P.; Shourie, A. Phyto-microbiome to mitigate abiotic stress in crop plants. Front. Microbiol. 2023, 14, 1210890. [Google Scholar]
- Dastogeer, K.M.G.; Zahan, M.I.; Tahjib-Ul-Arif, M.; Akter, M.A.; Okazaki, S. Plant salinity tolerance conferred by arbuscular mycorrhizal fungi and associated mechanisms: A meta-analysis. Front. Plant Sci. 2020, 11, 588550. [Google Scholar]
- Fu, H.; Yang, Y. How plants tolerate salt stress. Curr. Issues Mol. Biol. 2023, 45, 5914–5934. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Raghuvanshi, N.; Kumar, A.; Rithesh, L.; Rai, A.; Pandey, A.K. Deciphering phytomicrobiomes for sustainable crop production: Recent findings and future perspectives. Plant Stress 2023, 10, 100285. [Google Scholar] [CrossRef]
- Zeng, Q.; Man, X.; Dai, Y.C.; Liu, H. Pseudomonas Spp. Enriched in Endophytic Community of Healthy Cotton Plants Inhibit Cotton Verticillium Wilt. Front. Microbiol. 2022, 13, 906732. [Google Scholar]
- Wang, J. Leaf Phyllosphere Microbiome and Crop Productivity Under Climate Warming. Crop. Sci. 2023, 63, 987–989. [Google Scholar]
- Terhonen, E.; Blumenstein, K.; Kovalchuk, A.; Asiegbu, F.O. Forest tree microbiomes and associated fungal endophytes: Functional roles and impact on forest health. Forests 2019, 10, 42. [Google Scholar] [CrossRef]
- Adedayo, A.A.; Fadiji, A.E.; Babalola, O.O. Unraveling the functional genes present in rhizosphere microbiomes of Solanum lycopersicum. PeerJ 2023, 11, e15432. [Google Scholar]
- Anguita-Maeso, M.; Navas-Cortés, J.A.; Landa, B.B. Insights into the methodological, biotic and abiotic factors influencing the characterization of xylem-inhabiting microbial communities of olive trees. Plants 2023, 12, 912. [Google Scholar] [CrossRef] [PubMed]
- Romano, I.; Ventorino, V.; Pepe, O. Effectiveness of plant beneficial microbes: Overview of the methodological approaches for the assessment of root colonization and persistence. Front. Plant Sci. 2020, 11, 6. [Google Scholar] [CrossRef]
- Jing, J.; Garbeva, P.; Raaijmakers, J.M.; Medema, M.H. Strategies for tailoring functional microbial synthetic communities. ISME J. 2024, 18, wrae049. [Google Scholar]
- Wang, Z.; Hu, X.; Solanki, M.K.; Pang, F. A Synthetic Microbial Community of Plant Core Microbiome Can Be a Potential Biocontrol Tool. J. Agric. Food Chem. 2023, 71, 5030–5041. [Google Scholar]
- Trivedi, P.; Mattupalli, C.; Eversole, K.; Leach, J.E. Enabling sustainable agriculture through understanding and enhancement of microbiomes. New Phytol. 2021, 230, 2129–2147. [Google Scholar]
- Pereira-Flores, E.; Glöckner, F.O.; Fernandez-Guerra, A. Fast and accurate average genome size and 16S rRNA gene average copy number computation in metagenomic data. BMC Bioinform. 2019, 20, 453. [Google Scholar]
- McPherson, M.R.; Wang, P.; Marsh, E.L.; Mitchell, R.B.; Schachtman, D.P. Isolation and analysis of microbial communities in soil, rhizosphere, and roots in perennial grass experiments. J. Vis. Exp. 2018, 137, 57932. [Google Scholar]
- Fang, J.; Tao, Y.; Liu, J.; Lyu, T.; Yang, X.; Ma, S.; Dong, J.; Dou, H.; Zhang, H. Effects of emergent plants on soil carbon-fixation and denitrification processes in freshwater and brackish wetlands in a watershed in northern China. Geoderma 2023, 430, 116311. [Google Scholar] [CrossRef]
- Kandlikar, G.S.; Gold, Z.J.; Cowen, M.C.; Meyer, R.S.; Freise, A.C.; Kraft, N.J.; Moberg-Parker, J.; Sprague, J.; Kushner, D.J.; Curd, E.E. ranacapa: An R package and Shiny web app to explore environmental DNA data with exploratory statistics and interactive visualizations [version 1; referees: 1 approved, 2 approved with reservations]. F1000Research 2018, 7, 1734. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Gao, L.; Huang, Y.; Liu, Y.; Mohamed, O.A.A.; Fan, X.; Wang, L.; Li, L.; Ma, J. Bacterial community structure and potential microbial coexistence mechanism associated with three halophytes adapting to the extremely hypersaline environment. Microorganisms 2022, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, F.; Sedghi, M.; Pazuki, A.; Pessarakli, M. Investigation of seed germination indices for early selection of salinity tolerant genotypes: A case study in wheat. Emir. J. Food Agric. 2017, 29, 222–226. [Google Scholar]
- Stojšin, M.M.; Petrović, S.; Dimitrijević, M.; Malenčić, D.; Zečević, V.; Banjac, B.; Knežević, D. Effect of salinity stress on antioxidant activity and grain yield of different wheat genotypes. Turk. J. Field Crops 2022, 27, 33–40. [Google Scholar]
- Amin, A.; Ahmed, I.; Khalid, N.; Khan, I.U.; Ali, A.; Dahlawi, S.M.; Li, W.-J. Insights on comparative bacterial diversity between different arid zones of Cholistan Desert, Pakistan. 3 Biotech 2020, 10, 224. [Google Scholar]
- Yang, X.; Dai, Z.; Yuan, R.; Guo, Z.; Xi, H.; He, Z.; Wei, M. Effects of salinity on assembly characteristics and function of microbial communities in the phyllosphere and rhizosphere of salt-tolerant Avicennia marina mangrove species. Microbiol. Spectr. 2023, 11, e0300022. [Google Scholar] [PubMed]
- Wang, S.; Guo, Z.; Wang, L.; Zhang, Y.; Jiang, F.; Wang, X.; Yin, L.; Liu, B.; Liu, H.; Wang, H.; et al. Wheat rhizosphere metagenome reveals newfound potential soil Zn-mobilizing bacteria contributing to cultivars’ variation in grain Zn concentration. Front. Microbiol. 2021, 12, 689855. [Google Scholar]
- Hone, H.; Mann, R.; Yang, G.; Kaur, J.; Tannenbaum, I.; Li, T.; Spangenberg, G.; Sawbridge, T. Profiling, isolation and characterisation of beneficial microbes from the seed microbiomes of drought tolerant wheat. Sci. Rep. 2021, 11, 11916. [Google Scholar]
- Moore, E.R.; Suazo, D.; Babilonia, J.; Montoya, K.N.; Gallegos-Graves, L.V.; Sevanto, S.; Dunbar, J.; Albright, M.B.N. Drivers of Stability and Transience in Composition-Functioning Links During Serial Propagation of Litter-Decomposing Microbial Communities. mSystems 2023, 8, e01220-22. [Google Scholar]
- Shu, X. Ecosystem Multifunctionality and Soil Microbial Communities in Response to Ecological Restoration in an Alpine Degraded Grassland. Front. Plant Sci. 2023, 14, 1173962. [Google Scholar]
- Salimi, A.; Etemadi, M.; Eshghi, S.; Karami, A.; Alizargar, J. The effects of halomonas sp. and azotobacter sp. on ameliorating the adverse effect of salinity in purple basil (Ocimum basilicum L.). Preprints 2021, 2021, 2021060391. [Google Scholar]
- Yañez-Yazlle, M.F.; Romano-Armada, N.; Rajal, V.B.; Irazusta, V.P. Amelioration of saline stress on chia (Salvia hispanica L.) seedlings inoculated with halotolerant plant growth-promoting bacteria isolated from hypersaline environments. Front. Agron. 2021, 3, 665798. [Google Scholar] [CrossRef]
- Zhou, Y.; Hao, L.; Ji, C.; Zhou, Q.; Song, X.; Liu, Y.; Li, H.; Li, C.; Gao, Q.; Li, J.; et al. The Effect of Salt-Tolerant Antagonistic Bacteria CZ-6 on the Rhizosphere Microbial Community of Winter Jujube (Ziziphus Jujuba Mill. “Dongzao”) in Saline-Alkali Land. Biomed. Res. Int. 2021, 2021, 6296896. [Google Scholar] [CrossRef] [PubMed]
- Fathalla, A.; EL-Mageed, A. Salt Tolerance Enhancement of Wheat (Triticum asativium L.) Genotypes by Selected Plant Growth Promoting Bacteria. AIMS Microbiol. 2020, 6, 250–271. [Google Scholar]
- Pandey, S.; Gupta, S. Evaluation of Pseudomonas Sp. For Its Multifarious Plant Growth Promoting Potential and Its Ability to Alleviate Biotic and Abiotic Stress in Tomato (Solanum lycopersicum) Plants. Sci. Rep. 2020, 10, 20951. [Google Scholar]
- Safari, D.; Jamali, F.; Nooryazdan, H.; Bayat, F. Evaluation of ACC Deaminase Producing Pseudomonas Fluorescens Strains for Their Effects on Seed Germination and Early Growth of Wheat Under Salt Stress. Aust. J. Crop. Sci. 2018, 12, 413–421. [Google Scholar]
- Cappellari, L.D.R. Exploring the Differential Impact of Salt Stress on Root Colonization Adaptation Mechanisms in Plant Growth-Promoting Rhizobacteria. Plants 2023, 12, 4059. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Ali, Q.; Jiang, Q.; Wang, R.; Wang, Z.; Mu, G.; Khan, S.A.; Khan, A.R.; Manghwar, H.; Wu, H.; et al. Salt Tolerant Bacillus Strains Improve Plant Growth Traits and Regulation of Phytohormones in Wheat Under Salinity Stress. Plants 2022, 11, 2769. [Google Scholar] [CrossRef]
- Rauf, M.; Awais, M.; Ud-Din, A.; Ali, K.; Gul, H.; Rahman, M.M.; Hamayun, M.; Arif, M. Molecular Mechanisms of the 1-Aminocyclopropane-1-Carboxylic Acid (ACC) Deaminase Producing Trichoderma Asperellum MAP1 in Enhancing Wheat Tolerance to Waterlogging Stress. Front. Plant Sci. 2021, 11, 614971. [Google Scholar] [CrossRef]
- Zaheer, M.S.; Ali, H.H.; Iqbal, M.A.; Erinle, K.O.; Javed, T.; Iqbal, J.; Hashmi, M.I.U.; Mumtaz, M.Z.; Salama, E.A.A.; Kalaji, H.M.; et al. Cytokinin Production by Azospirillum Brasilense Contributes to Increase in Growth, Yield, Antioxidant, and Physiological Systems of Wheat (Triticum aestivum L.). Front. Microbiol. 2022, 13, 886041. [Google Scholar]
- Jacob, S.; Sudini, H.K. Indirect plant growth promotion in grain legumes: Role of actinobacteria. In Plant Growth Promoting Actinobacteria: A New Avenue for Enhancing the Productivity and Soil Fertility of Grain Legumes; Springer: Singapore, 2016; pp. 17–32. [Google Scholar]
- Li, X.; Zheng, J.; Wei, W.; Gong, Z.; Liu, Z. The halophilic bacteria Gracilibacillus dipsosauri GDHT17 alleviates salt stress on perennial ryegrass seedlings. Front. Microbiol. 2023, 14, 1213884. [Google Scholar]
- Chakraborty, U.; Roy, S.; Chakraborty, B. Microorganisms aiding existence and efficiency of plants in saline environment: What we know and what to expect. In Microorganisms in Saline Environments: Strategies and Functions; Springer: Cham, Switzerland, 2019; pp. 211–235. [Google Scholar]
- Teotia, N.; Chaudhary, D.R. Application of halophyte microbiome for development of salt tolerance in crops. In Improving Stress Resilience in Plants; Elsevier: Amsterdam, The Netherlands, 2024; pp. 143–164. [Google Scholar]
- Hussein, K.A.; Yoo, J.H.; Joo, J.H. Tolerance to Salt Stress by Plant Growth-Promoting Rhizobacteria on Brassica Rapa Var. Glabra. Korean J. Soil Sci. Fertil. 2016, 49, 776–782. [Google Scholar] [CrossRef]
- Xu, Y.; Ge, Y.; Song, J.; Rensing, C. Assembly of root-associated microbial community of typical rice cultivars in different soil types. Biol. Fertil. Soils 2020, 56, 249–260. [Google Scholar] [CrossRef]
- Gonçalves, A.C.; Sánchez-Juanes, F.; Meirinho, S.; Silva, L.R.; Alves, G.; Flores-Félix, J.D. Insight into the taxonomic and functional diversity of bacterial communities inhabiting blueberries in Portugal. Microorganisms 2022, 10, 2193. [Google Scholar] [CrossRef]
- Jespersen, D.; Yu, J.; Huang, B. Metabolic Effects of Acibenzolar-S-Methyl for Improving Heat or Drought Stress in Creeping Bentgrass. Front. Plant Sci. 2017, 8, 1224. [Google Scholar]
- Almeida, O.A.C.; de Araujo, N.O.; Mulato, A.T.N.; Persinoti, G.F.; Sforça, M.L.; Calderan-Rodrigues, M.J.; Oliveira, J.V.d.C. Bacterial Volatile Organic Compounds (VOCs) Promote Growth and Induce Metabolic Changes in Rice. Front. Plant Sci. 2023, 13, 1056082. [Google Scholar] [CrossRef]
- Barghi, A. Bacterial Volatile Organic Compounds From Bacillus Zanthoxyli HS-1 Enhance Tolerance Responses Against Salt and High Temperature in Cucumber and Cabbage. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Khairunisa, B.H.; Susanti, D.; Loganathan, U.; Teutsch, C.D.; Campbell, B.T.; Fiske, D.; Wilkinson, C.A.; Aylward, F.O.; Mukhopadhyay, B. Dominant remodelling of cattle rumen microbiome by Schedonorus arundinaceus (tall fescue) KY-31 carrying a fungal endophyte. Access Microbiol. 2022, 4, 000322. [Google Scholar]
- Biasato, I.; Rimoldi, S.; Caimi, C.; Oddon, S.B.; Chemello, G.; Prearo, M.; Saroglia, M.; Hardy, R.; Gasco, L.; Terova, G. Efficacy of utilization of all-plant-based and commercial low-fishmeal feeds in two divergently selected strains of rainbow trout (Oncorhynchus mykiss): Focus on growth performance, whole-body proximate composition, and intestinal microbiome. Front. Physiol. 2022, 13, 892550. [Google Scholar] [CrossRef]
- Carlos, F.S.; Schaffer, N.; Marcolin, E.; Fernandes, R.S.; Mariot, R.; Mazzurana, M.; Roesch, L.F.W.; Levandoski, B.; Camargo, F.A. A Long-term No-tillage System Can Increase Enzymatic Activity and Maintain Bacterial Richness in Paddy Fields. Land Degrad. Dev. 2021, 32, 2257–2268. [Google Scholar] [CrossRef]
- Hirpara, K.R. Metagenomic Evaluation of Peanut Rhizosphere Microbiome From the Farms of Saurashtra Regions of Gujarat, India. Sci Rep. 2024, 14, 10525. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Counts |
|---|---|
| OTU annotation: | GreengenesID |
| OTU number: | 1110 |
| OTUs with ≥2 counts: | 752 |
| Number of experimental factors with replicates: | 3 [discrete: 3 continuous: 0] |
| Total read counts: | 4,526,902 |
| Average counts per sample: | 377,241 |
| Maximum counts per sample: | 796,235 |
| Minimum counts per sample: | 141,355 |
| Number of sample names matched (metadata vs. OTU table): | 12 |
| Plant–Microbial Interaction (Microbial Recruitment) | ||||||
|---|---|---|---|---|---|---|
| No. | Phylum (Class) | Taxa | Log2FC | Estimated BS | Fold Change | Percentage Increase (%) |
| 1 | Proteobacteria (DeltaProteobacteria) | Sandaracinaceae | 3.37 | 0.097 | 10.34 | 933.88 |
| 2 | Proteobacteria (GammaProteobacteria) | Halomonas_xianhensis | 3.83 | 0.07 | 14.22 | 1322.15 |
| 3 | Legionella | 3.64 | 0.08 | 12.47 | 1146.66 | |
| 4 | Thermomonas | 3.58 | 0.084 | 11.96 | 1095.88 | |
| 5 | Azotobacter_armeniacus | 3.28 | 0.103 | 9.71 | 871.36 | |
| 6 | Rhizobacter | 3.14 | 0.113 | 8.82 | 781.52 | |
| 7 | Pseudomonas_sagittaria | 2.87 | 0.137 | 7.31 | 631.06 | |
| 8 | Rhizobacter_fulvus | 2.78 | 0.145 | 6.87 | 586.85 | |
| 9 | Halomonas_daqiaonensis | 2.43 | 0.185 | 5.39 | 438.89 | |
| 10 | Rheinheimera | 2.35 | 0.196 | 5.1 | 409.82 | |
| 11 | Pseudomonas_azotifigens | 2.34 | 0.198 | 5.06 | 405.88 | |
| 12 | Proteobacteria (AlphaProteobacteria) | Devosia_terrae | 2.77 | 0.147 | 6.82 | 582.11 |
| 13 | Altererythrobacter_sp_ | 2.22 | 0.215 | 4.66 | 366.24 | |
| 14 | Novosphingobium | 2.11 | 0.231 | 4.32 | 331.69 | |
| 15 | Altererythrobacter | 2.09 | 0.234 | 4.26 | 326.27 | |
| 16 | Actinobacteria | Saccharothrix_hoggarensis | 3.46 | 0.091 | 11 | 1000.43 |
| 17 | Saccharothrix | 4.03 | 0.061 | 16.34 | 1533.62 | |
| 18 | Firmicutes (Bacilli) | Gracilibacillus_ureilyticus | 3.26 | 0.104 | 9.58 | 858.35 |
| 19 | Fibrobacteres (Fibrobacteria) | Fibrobacteraceae | 2.29 | 0.205 | 4.89 | 388.93 |
| 20 | Bacteroidetes (Flavobacteriia) | Ulvibacter | 2.81 | 0.143 | 7.01 | 601.28 |
| 21 | Verrucomicrobia (Opitutae) | Opitutaceae | 2.11 | 0.231 | 4.32 | 331.69 |
| 22 | Puniceicoccaceae | 2.24 | 0.212 | 4.72 | 372.43 | |
| Salinity-Induced Microbial Shifts | ||||||
|---|---|---|---|---|---|---|
| No. | Phylum (Class) | Taxa | Log2FC | RC | Fold Change (FC) | Percentage Increase of RS over RC (%) |
| 1 | Proteobacteria (GammaProteobacteria) | Idiomarinaceae | 6.11 | 0.014477938 | 69.07060714 | 6807.060714 |
| 2 | Microbulbifer | 3.61 | 0.081899588 | 12.21007367 | 1121.007367 | |
| 3 | Rheinheimera | 3.43 | 0.092782723 | 10.77786861 | 977.7868615 | |
| 4 | Halomonadaceae | 3.13 | 0.114228931 | 8.75434961 | 775.434961 | |
| 5 | Marinobacter_sp_ | 3.11 | 0.115823508 | 8.633825892 | 763.3825892 | |
| 6 | Halomonas | 2.98 | 0.126744935 | 7.889861636 | 688.9861636 | |
| 7 | Marinobacter | 2.8 | 0.143587294 | 6.964404506 | 596.4404506 | |
| 8 | Pseudomonas_sp_ | 2.29 | 0.204475515 | 4.890561111 | 389.0561111 | |
| 9 | Pseudomonas_sagittaria | 2.17 | 0.22221067 | 4.500233939 | 350.0233939 | |
| 10 | Firmicutes (Bacilli) | Gracilibacillus | 4.45 | 0.045752678 | 21.85664411 | 2085.664411 |
| 11 | Gracilibacillus_kekensis | 3.73 | 0.075362989 | 13.26911273 | 1226.911273 | |
| 12 | Bacillaceae | 2.04 | 0.243163737 | 4.112455307 | 311.2455307 | |
| 13 | Bacteroidetes (Cytophagia) | Nafulsella | 3.72 | 0.07588718 | 13.17745628 | 1217.745628 |
| 14 | Bacteroidetes (Flavobacteriia) | Salegentibacter | 3.71 | 0.076415017 | 13.08643294 | 1208.643294 |
| 15 | Salinimicrobium_marinum | 3.45 | 0.091505356 | 10.92832205 | 992.8322054 | |
| 16 | Psychroflexus | 2.86 | 0.137738139 | 7.260153243 | 626.0153243 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fouad, N.; El-Zayat, E.M.; Amr, D.; El-Khishin, D.A.; Abd-Elhalim, H.M.; Hafez, A.; Radwan, K.H.; Hamwieh, A.; Tadesse, W. Characterizing Wheat Rhizosphere Bacterial Microbiome Dynamics Under Salinity Stress: Insights from 16S rRNA Metagenomics for Enhancing Stress Tolerance. Plants 2025, 14, 1033. https://doi.org/10.3390/plants14071033
Fouad N, El-Zayat EM, Amr D, El-Khishin DA, Abd-Elhalim HM, Hafez A, Radwan KH, Hamwieh A, Tadesse W. Characterizing Wheat Rhizosphere Bacterial Microbiome Dynamics Under Salinity Stress: Insights from 16S rRNA Metagenomics for Enhancing Stress Tolerance. Plants. 2025; 14(7):1033. https://doi.org/10.3390/plants14071033
Chicago/Turabian StyleFouad, Nourhan, Emad M. El-Zayat, Dina Amr, Dina A. El-Khishin, Haytham M. Abd-Elhalim, Amr Hafez, Khaled H. Radwan, Aladdin Hamwieh, and Wuletaw Tadesse. 2025. "Characterizing Wheat Rhizosphere Bacterial Microbiome Dynamics Under Salinity Stress: Insights from 16S rRNA Metagenomics for Enhancing Stress Tolerance" Plants 14, no. 7: 1033. https://doi.org/10.3390/plants14071033
APA StyleFouad, N., El-Zayat, E. M., Amr, D., El-Khishin, D. A., Abd-Elhalim, H. M., Hafez, A., Radwan, K. H., Hamwieh, A., & Tadesse, W. (2025). Characterizing Wheat Rhizosphere Bacterial Microbiome Dynamics Under Salinity Stress: Insights from 16S rRNA Metagenomics for Enhancing Stress Tolerance. Plants, 14(7), 1033. https://doi.org/10.3390/plants14071033