
Identification and Characterization of Copper-Responsive miRNAs and Their Target Genes in Jerusalem Artichoke
Abstract
1. Introduction
2. Results
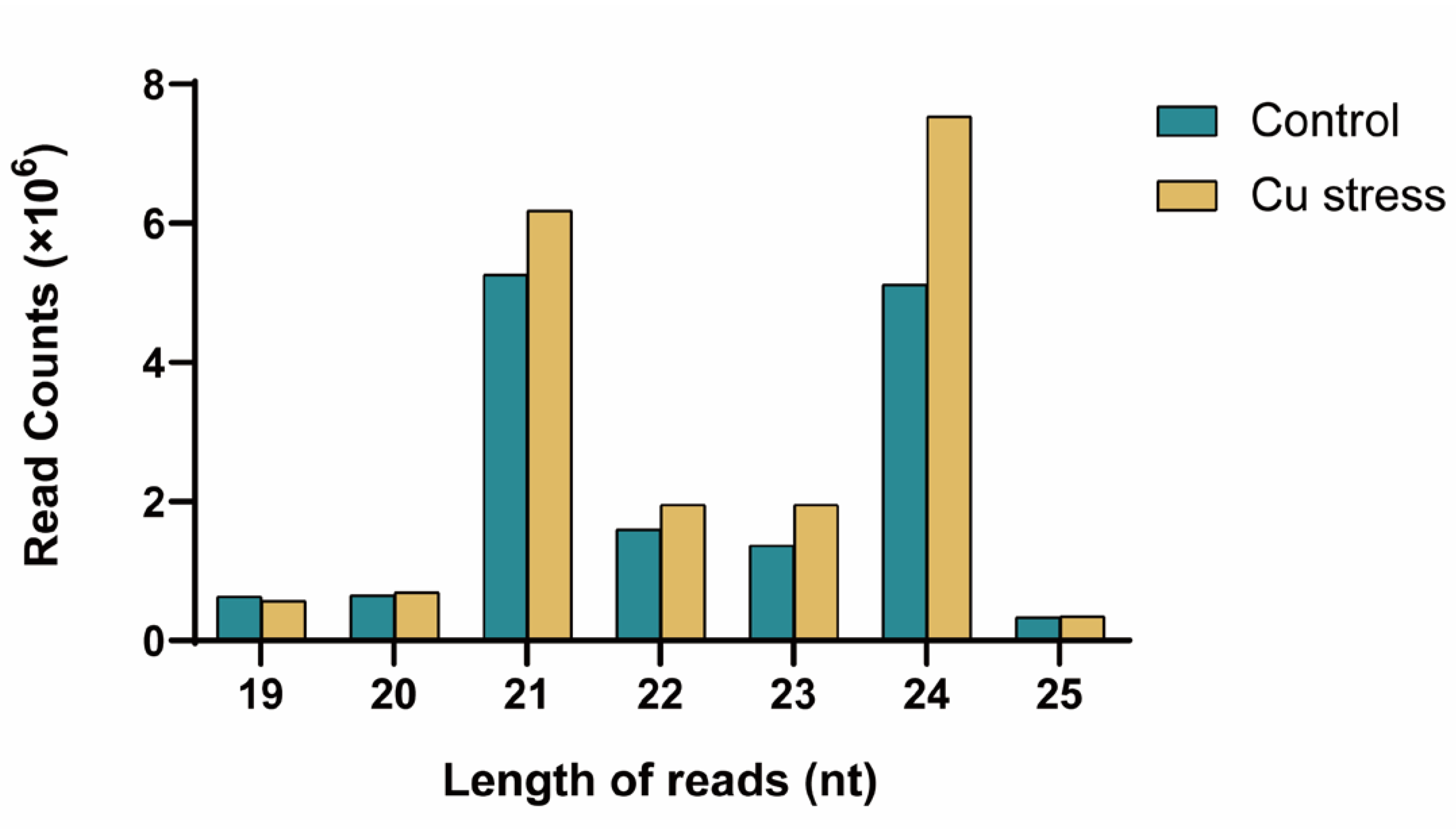
2.1. Analysis of Small RNA Sequencing Data
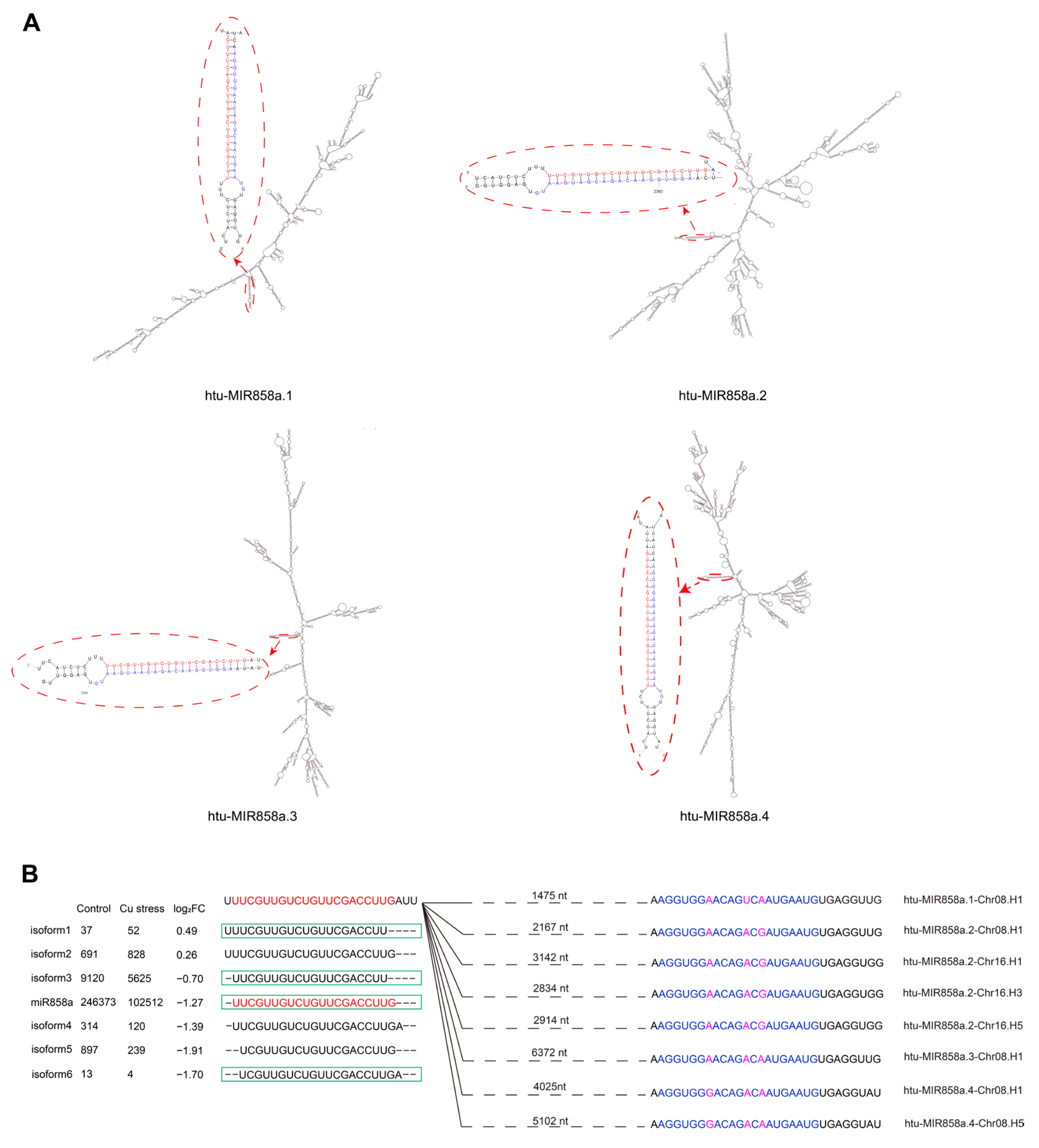
2.2. Identification and Characterization of Differentially Expressed miRNAs in Two Libraries
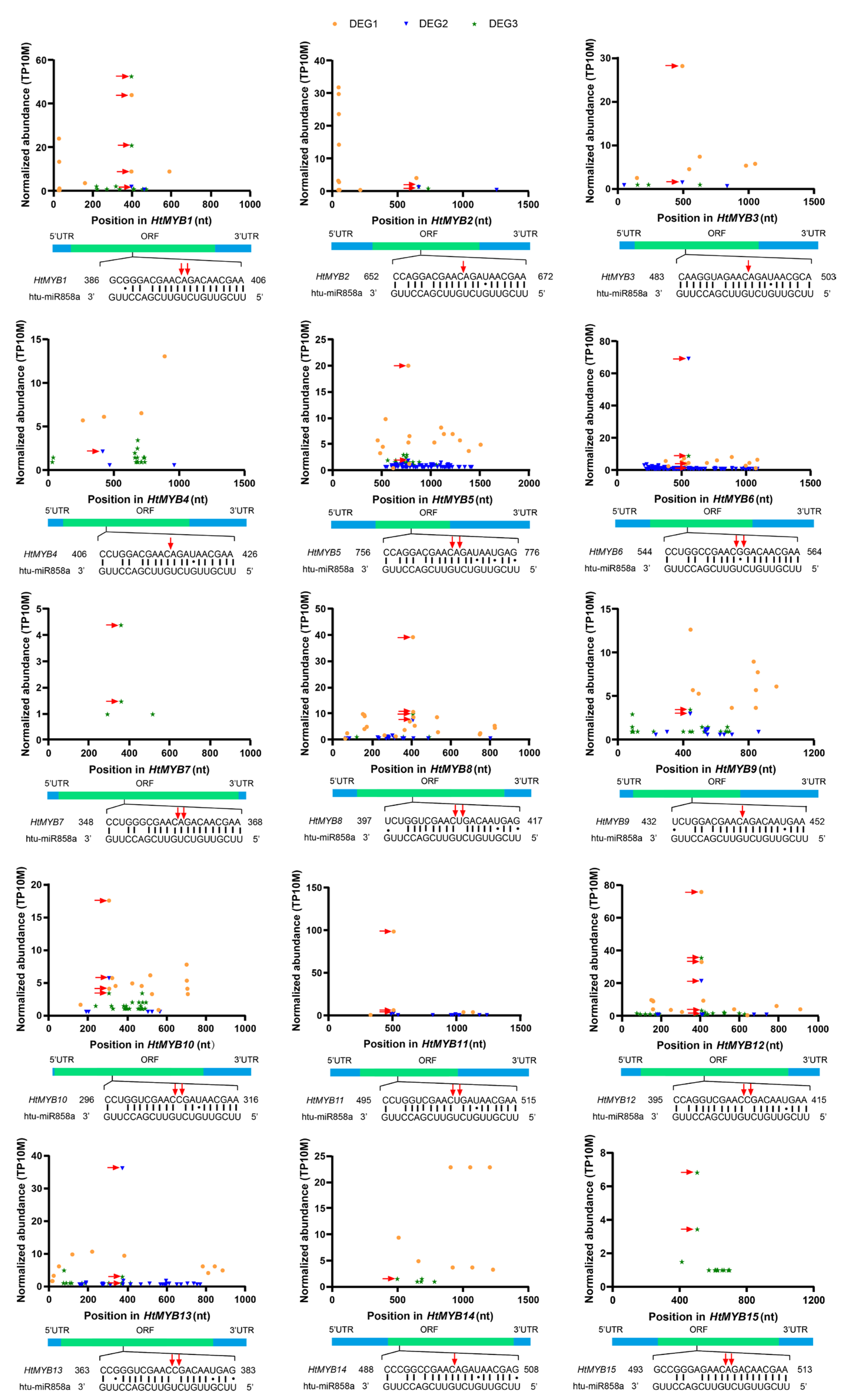
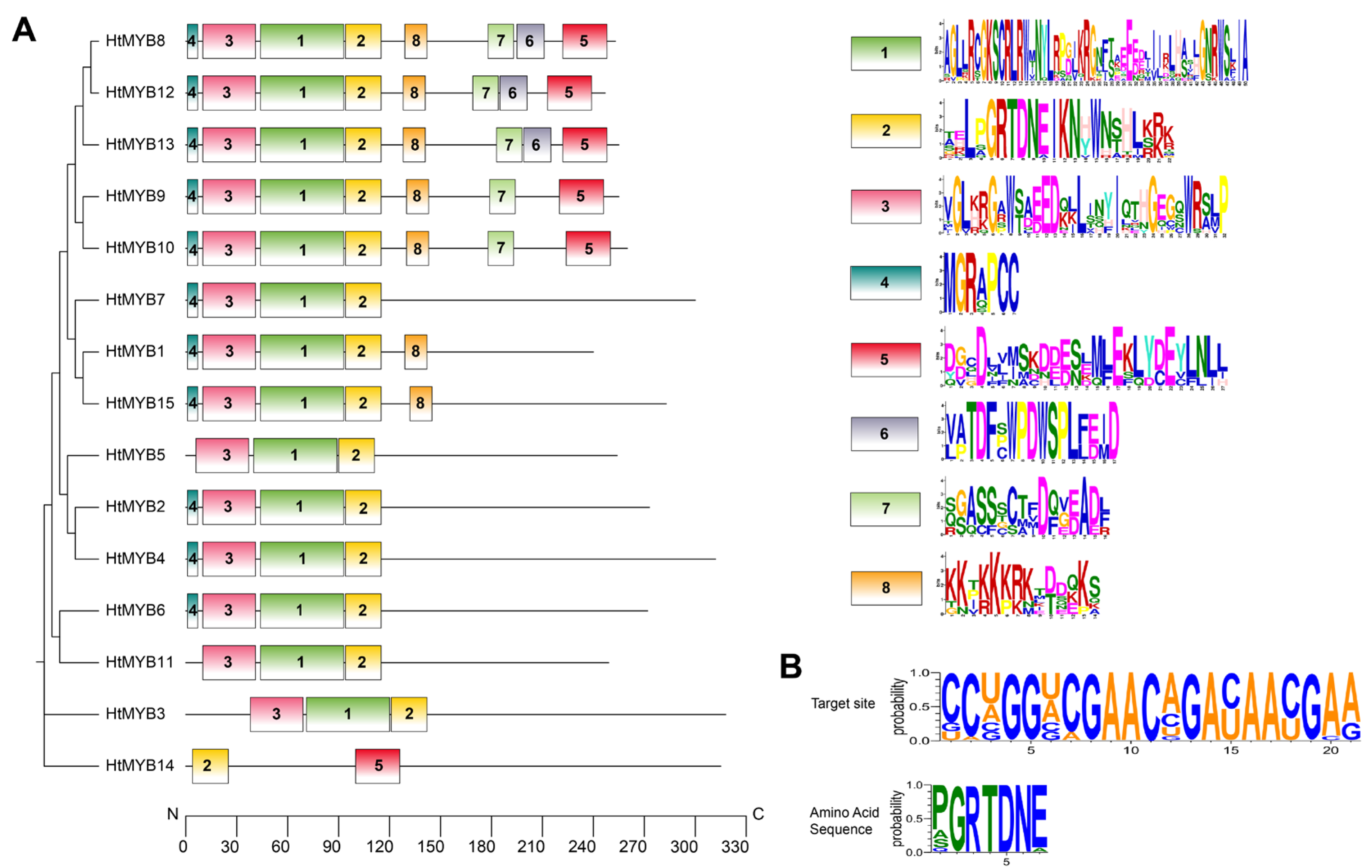
2.3. Identification and Characterization of miRNA Targets
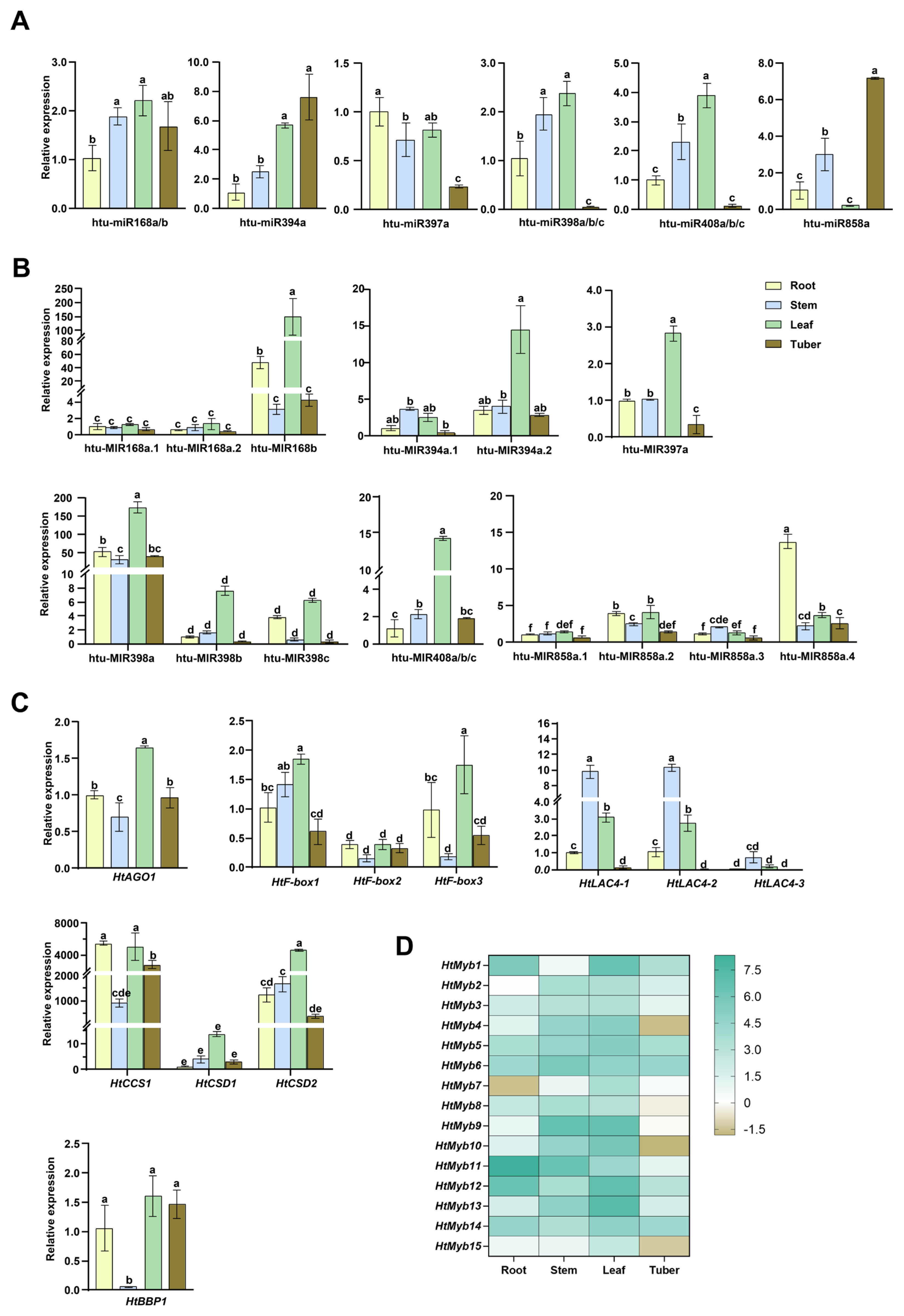
2.4. Expression Levels of miRNAs and Target Genes Across Different Tissues
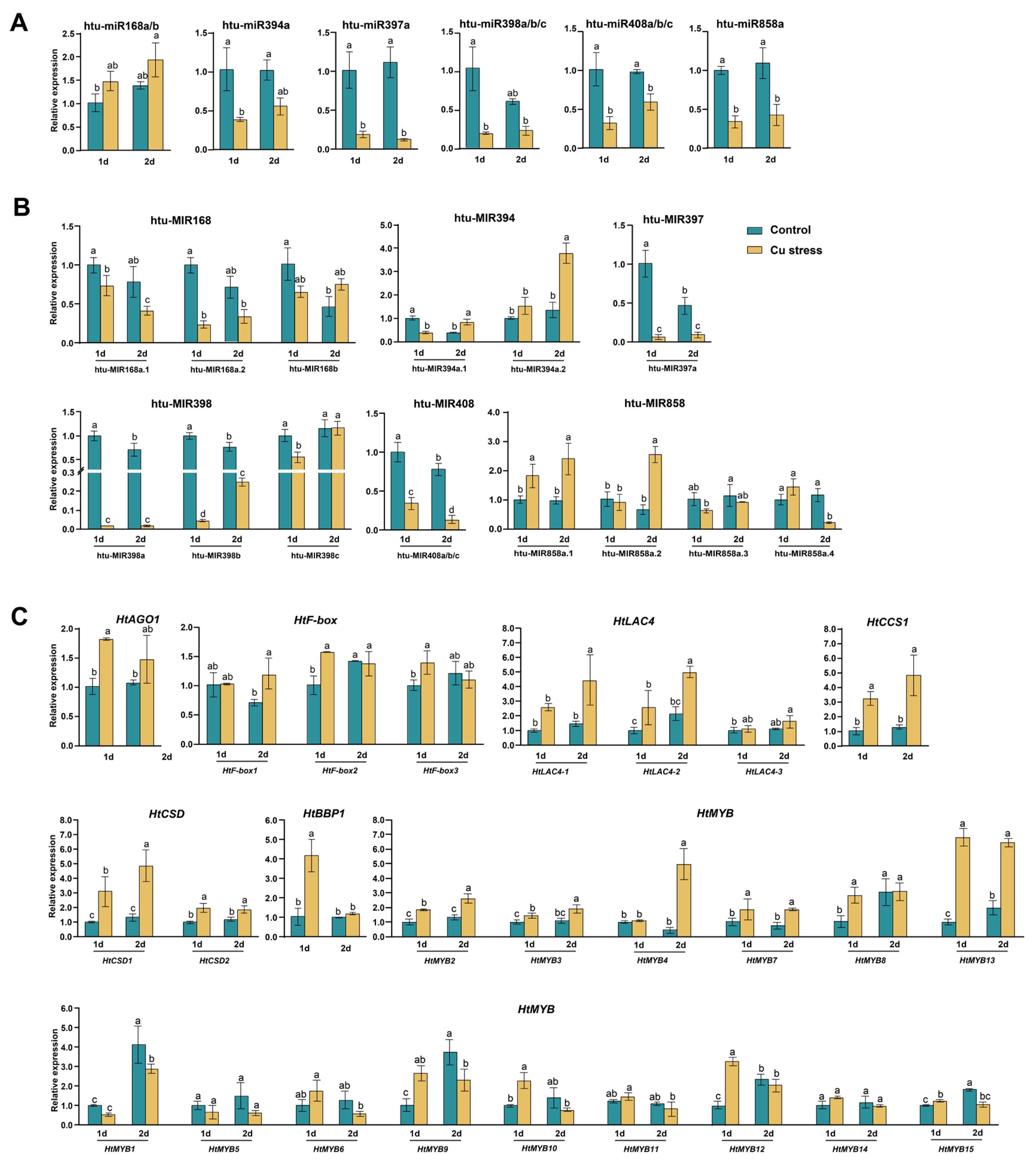
2.5. Expression Analysis of miRNAs and Their Target Genes Under Cu Stress
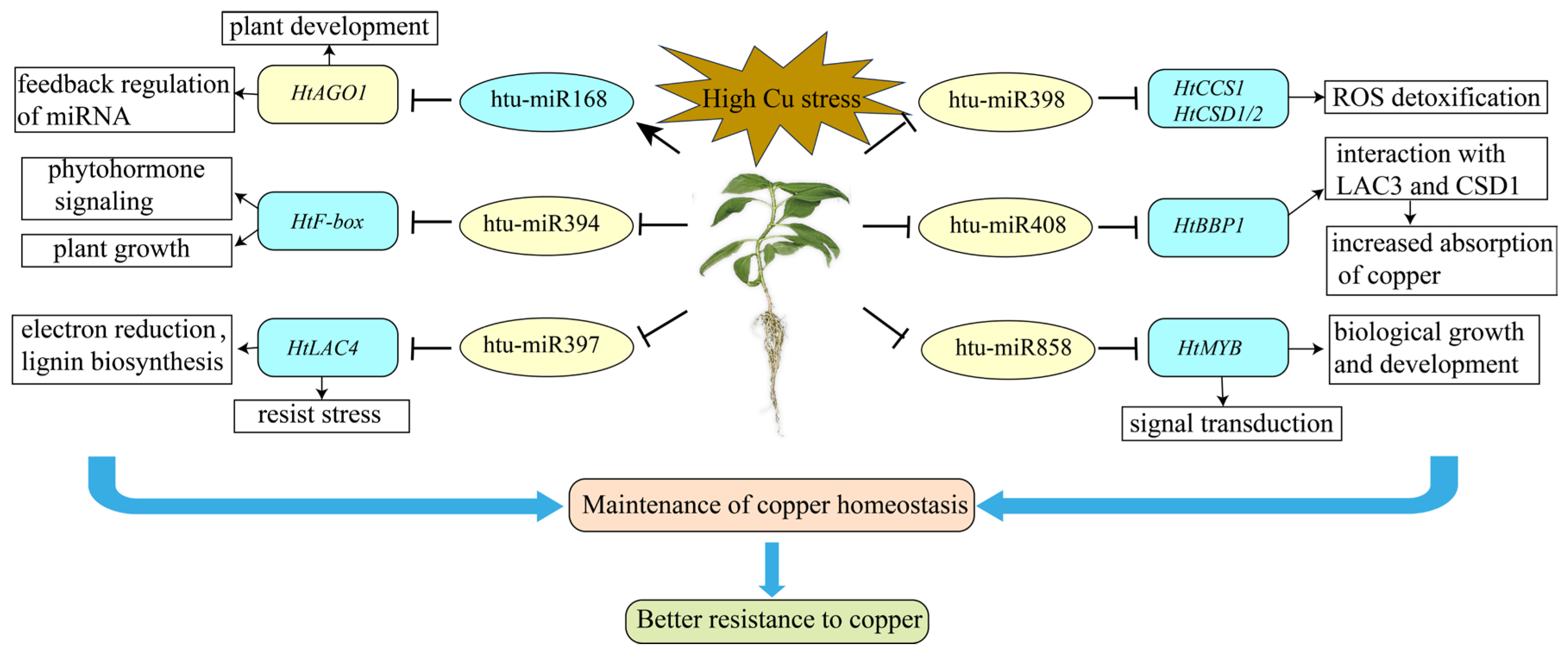
3. Discussion
4. Materials and Methods
4.1. Plant Culture and Treatment
4.2. RNA Extraction and sRNA Library Construction and Sequencing
4.3. sRNA-seq Data Processing and miRNA Identification
4.4. Target Identification and Other Bioinformatics Analyses
4.5. cDNA Synthesis and Gene Cloning and Expression
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Cu | Copper |
| miRNA | microRNA |
| qRT-PCR | quantitative real-time PCR |
| AGO | Argonaute |
| LAC | Laccase |
| CCS | Copper chaperone for superoxide dismutase |
| CSD | Copper/Zinc superoxide dismutase |
| BBP | Basic blue protein |
| MYB | Myeloblastosis |
| RISC | RNA-induced silencing complex |
| ORF | Open reading frame |
| AA | Amino acids |
| MW | Molecular weight |
| pI | Isoelectric point |
| GRAVY | Grand average of hydropathicity |
| nt | nucleotide |
References
- Wang, R.X.; Wang, Z.H.; Sun, Y.D.; Wang, L.L.; Li, M.; Liu, Y.T.; Zhang, H.M.; Jing, P.W.; Shi, Q.F.; Yu, Y.H. Molecular mechanism of plant response to copper stress: A review. Environ. Exp. Bot. 2024, 218, 105590. [Google Scholar] [CrossRef]
- Xu, E.; Liu, Y.; Gu, D.; Zhan, X.; Li, J.; Zhou, K.; Zhang, P.; Zou, Y. Molecular mechanisms of plant responses to copper: From deficiency to excess. Int. J. Mol. Sci. 2024, 25, 6993. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, F.; Azenha, M.; Silva, A.F.; de Sousa, A.; Santiago, A.; Ferraz, P.; Teixeira, J. Copper-induced stress in Solanum nigrum L. and antioxidant defense system responses. Food Energy Secur. 2013, 2, 70–80. [Google Scholar] [CrossRef]
- Kumar, V.; Pandita, S.; Singh Sidhu, G.P.; Sharma, A.; Khanna, K.; Kaur, P.; Bali, A.S.; Setia, R. Copper bioavailability, uptake, toxicity and tolerance in plants: A comprehensive review. Chemosphere 2021, 262, 127810. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Cho, S.W.; Kwon, S.J.; Kamal, A.H.M.; Lee, D.G.; Sarker, K.; Lee, M.S.; Xin, Z.G.; Woo, S.H. Proteome characterization of copper stress responses in the roots of sorghum. Biometals 2017, 30, 765–785. [Google Scholar] [CrossRef]
- Dubey, S.; Shri, M.; Gupta, A.; Rani, V.; Chakrabarty, D. Toxicity and detoxification of heavy metals during plant growth and metabolism. Environ. Chem. Lett. 2018, 16, 1169–1192. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, F.; Liu, J.L.; Wu, H.T.; Yang, H.; Shi, Y.; Liu, J.; Zhang, Y.F.; Luo, Y.R.; Chen, K.M. Heavy metal transporters: Functional mechanisms, regulation, and application in phytoremediation. Sci. Total Environ. 2022, 809, 151099. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, X.; Chen, J. Advances of the mechanism for copper tolerance in plants. Plant Sci. 2025, 350, 112299. [Google Scholar] [CrossRef]
- Liu, H.F.; Xue, X.J.; Yu, Y.; Xu, M.M.; Lu, C.C.; Meng, X.L.; Zhang, B.G.; Ding, X.H.; Chu, Z.H. Copper ions suppress abscisic acid biosynthesis to enhance defence against Phytophthora infestans in potato. Mol. Plant Pathol. 2020, 2, 636–651. [Google Scholar] [CrossRef]
- Agarwal, P.; Mitra, M.; Banerjee, S.; Roy, S. MYB4 transcription factor, a member of R2R3—Subfamily of MYB domain protein, regulates cadmium tolerance via enhanced protection against oxidative damage and increases expression of PCS1 and MT1C in Arabidopsis. Plant Sci. 2020, 297, 110501. [Google Scholar] [CrossRef]
- Ding, J.; Ji, C.; Yu, L.; Wang, C.; Ding, G.; Wang, S.; Shi, L.; Xu, F.; Cai, H. OsMYB84, a transcriptional regulator of OsCOPT2 and OsHMA5, modulates copper uptake and transport and yield production in rice. Crop J. 2024, 12, 456–469. [Google Scholar] [CrossRef]
- Xie, Y.F.; Zhang, R.X.; Qin, L.J.; Song, L.L.; Zhao, D.G.; Xia, Z.M. Genome-wide identification and genetic characterization of the CaMYB family and its response to five types of heavy metal stress in hot pepper (Capsicum annuum cv. CM334). Plant Physiol. Biochem. 2022, 170, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Ai, T.N.; Naing, A.H.; Yun, B.W.; Lim, S.H.; Kim, C.K. Overexpression of RsMYB1 enhances anthocyanin accumulation and heavy metal stress tolerance in transgenic petunia. Front. Plant Sci. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Sunkar, R.; Kapoor, A.; Zhu, J.K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006, 18, 2051–2065. [Google Scholar] [CrossRef]
- Yamasaki, H.; Abdel-Ghany, S.E.; Cohu, C.M.; Kobayashi, Y.; Shikanai, T.; Pilon, M. Regulation of copper homeostasis by micro-RNA in Arabidopsis. J. Biol. Chem. 2007, 282, 16369–16378. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Pilon, M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem. 2008, 283, 15932–15945. [Google Scholar] [CrossRef]
- Leng, X.; Wang, P.; Zhao, P.; Wang, M.; Cui, L.; Shangguan, L.; Wang, C. Conservation of microRNA-mediated regulatory networks in response to copper stress in grapevine. Plant Growth Regul. 2017, 82, 293–304. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, X.; Li, J.; Cai, H.; Deng, X.W.; Li, L. MicroRNA408 is critical for the HY5-SPL7 gene network that mediates the coordinated response to light and copper. Plant Cell. 2014, 26, 4933–4953. [Google Scholar] [CrossRef]
- Shahbaz, M.; Pilon, M. Conserved Cu-microRNAs in Arabidopsis thaliana function in copper economy under deficiency. Plants 2019, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhong, Q.; Tian, J.; Wang, L.; Zhao, M.; Li, L.; Sun, X. Characterization and development of EST-SSR markers to study the genetic diversity and populations analysis of Jerusalem artichoke (Helianthus tuberosus L.). Genes Genom. 2018, 40, 1023–1032. [Google Scholar] [CrossRef]
- Long, X.H.; Shao, H.B.; Liu, L.; Liu, L.P.; Liu, Z.P. Jerusalem artichoke: A sustainable biomass feedstock for biorefinery. Renew. Sust. Energy Rev. 2016, 54, 1382–1388. [Google Scholar] [CrossRef]
- Fleming, S.E.; GrootWassink, J.W. Preparation of high-fructose syrup from the tubers of the Jerusalem artichoke (Helianthus tuberosus L.). CRC Crit. Rev. Food Sci. Nutr. 1979, 12, 1–28. [Google Scholar] [CrossRef]
- Lv, S.; Wang, R.; Xiao, Y.; Li, F.; Mu, Y.; Lu, Y.; Gao, W.; Yang, B.; Kou, Y.; Zeng, J.; et al. Growth, yield formation, and inulin performance of an on-food energy crop, Jerusalem artichoke (Helianthus tuberosus L.), in a semi-arid area of China. Ind. Crops Prod. 2019, 134, 71–79. [Google Scholar] [CrossRef]
- Long, X.; Ni, N.; Wang, L.; Wang, X.; Wang, J.; Zhang, Z.; Zed, R.; Liu, Z.; Shao, H. Phytoremediation of cadmium-contaminated soil by two Jerusalem Artichoke (Helianthus tuberosus L.) genotypes. Clean-Soil Air Water 2013, 41, 202–209. [Google Scholar] [CrossRef]
- Wen, F.L.; Yue, Y.; He, T.F.; Gao, X.M.; Zhou, Z.S.; Long, X.H. Identification of miR390-TAS3-ARF pathway in response to salt stress in Helianthus tuberosus L. Gene 2020, 738, 144460. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yue, Y.; Chen, X.; Long, X.; Zhou, Z. Identification and expression analysis of miR396 and its target genes in Jerusalem artichoke under temperature stress. Gene 2024, 893, 147908. [Google Scholar] [CrossRef]
- Chávez Montes, R.A.; de Fátima Rosas-Cárdenas, F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, N.; Meyers, B.C.; Green, P.J.; de Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef]
- Lunardon, A.; Johnson, N.R.; Hagerott, E.; Phifer, T.; Polydore, S.; Coruh, C.; Axtell, M.J. Integrated annotations and analyses of small RNA-producing loci from 47 diverse plants. Genome Res. 2020, 30, 497–513. [Google Scholar] [CrossRef]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant microRNA annotation in the era of big data. Plant Cell. 2018, 30, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Liu, X.F.; Zhu, Y.J.; Zhu, J.Z.; Liu, C.; Wang, Z.Y.; Shen, X.X.; Allan, A.C.; Yin, X.R. Identification of miRNA858 long-loop precursors in seed plants. Plant Cell. 2024, 36, 1637–1654. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, A.; Chen, R.; Xu, D.; Wang, H.; Jiang, F.; Liu, H.; Qian, W.; Fan, W. Haplotype-resolved chromosome-level genome of hexaploid Jerusalem artichoke provides insights into its origin, evolution, and inulin metabolism. Plant Commun. 2024, 5, 100767. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef]
- Zhou, Z.S.; Song, J.B.; Yang, Z.M. Genome-wide identification of Brassica napus microRNAs and their targets in response to cadmium. J. Exp. Bot. 2012, 63, 4597–4613. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef]
- Barozai, M.Y.; Baloch, I.A.; Din, M. Identification of MicroRNAs and their targets in Helianthus. Mol. Biol. Rep. 2012, 39, 2523–2532. [Google Scholar] [CrossRef]
- Monavar Feshani, A.; Mohammadi, S.; Frazier, T.P.; Abbasi, A.; Abedini, R.; Karimi Farsad, L.; Ehya, F.; Salekdeh, G.H.; Mardi, M. Identification and validation of Asteraceae miRNAs by the expressed sequence tag analysis. Gene 2012, 493, 253–259. [Google Scholar] [CrossRef]
- Thakur, V.; Wanchana, S.; Xu, M.; Bruskiewich, R.; Quick, W.P.; Mosig, A.; Zhu, X.G. Characterization of statistical features for plant microRNA prediction. BMC Genom. 2011, 12, 108. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Bajczyk, M.; Jarmolowski, A.; Jozwiak, M.; Pacak, A.; Pietrykowska, H.; Sierocka, I.; Swida-Barteczka, A.; Szewc, L.; Szweykowska-Kulinska, Z. Recent insights into plant miRNA biogenesis: Multiple layers of miRNA level regulation. Plants 2023, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.F.; Zhu, C. The role of microRNAs in copper and cadmium homeostasis. Biochem. Biophys. Res. Commun. 2009, 386, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, W.; Shen, H.; Zhu, X.; Zhai, L.; Xu, L.; Wang, R.; Gong, Y.; Limera, C.; Liu, L. Identification of radish (Raphanus sativus L.) miRNAs and their target genes to explore miRNA-mediated regulatory networks in lead (Pb) stress responses by high-throughput sequencing and degradome analysis. Plant Mol. Biol. Rep. 2015, 33, 358–376. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, J.; Sun, Q.; Wang, J.; Huang, L.; Fu, S.; Qin, S.; Xie, X.; Ge, S.; Li, X.; et al. microRNAs: Key players in plant response to metal toxicity. Int. J. Mol. Sci. 2022, 23, 8642. [Google Scholar] [CrossRef]
- Perea-García, A.; Andrés-Bordería, A.; Huijser, P.; Peñarrubia, L. The copper-microRNA pathway is integrated with developmental and environmental stress responses in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 22, 9547. [Google Scholar] [CrossRef]
- Jin, Q.; Xue, Z.; Dong, C.; Wang, Y.; Chu, L.; Xu, Y. Identification and characterization of microRNAs from tree peony (Paeonia ostii) and their response to copper stress. PLoS ONE 2015, 10, e0117584. [Google Scholar] [CrossRef]
- Jiu, S.; Leng, X.; Haider, M.S.; Dong, T.; Guan, L.; Xie, Z.; Li, X.; Shangguan, L.; Fang, J. Identification of copper (Cu) stress-responsive grapevine microRNAs and their target genes by high-throughput sequencing. R. Soc. Open Sci. 2019, 6, 180735. [Google Scholar] [CrossRef]
- Lu, S.; Yang, C.; Chiang, V.L. Conservation and diversity of microRNA-associated copper-regulatory networks in Populus trichocarpa. J. Integr. Plant Biol. 2011, 53, 879–891. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell. 2004, 14, 787–799. [Google Scholar] [CrossRef]
- Huang, S.; Zhou, J.; Gao, L.; Tang, Y. Plant miR397 and its functions. Funct. Plant Biol. 2021, 48, 361–370. [Google Scholar] [CrossRef]
- Feng, Y.Z.; Yu, Y.; Zhou, Y.F.; Yang, Y.W.; Lei, M.Q.; Lian, J.P.; He, H.; Zhang, Y.C.; Huang, W.; Chen, Y.Q. A natural variant of miR397 mediates a feedback loop in circadian rhythm. Plant Physiol. 2020, 182, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Gao, C.; Zhang, H.; Wen, F.; Tao, L.; Fu, G.; Xiong, J. The evolution and functional roles of miR408 and its targets in plants. Int. J. Mol. Sci. 2022, 23, 530. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Ji, J.; Cheng, H.; Luo, R.; Zhang, J.; Li, W.; Wang, X.; Zhang, J.; Yao, Y. The miR408a-BBP-LAC3/CSD1 module regulates anthocyanin biosynthesis mediated by crosstalk between copper homeostasis and ROS homeostasis during light induction in Malus plants. J. Adv. Res. 2023, 51, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Denli, A.M.; Hannon, G.J. Biochemical specialization within Arabidopsis RNA silencing pathways. Mol. Cell. 2005, 19, 421–428. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, R.; Jia, X.; Tang, X.; Guo, Y.; Yang, H.; Zheng, X.; Qian, Q.; Qi, Y.; Zhang, Y. CRISPR-Cas9 mediated OsMIR168a knockout reveals its pleiotropy in rice. Plant Biotechnol. J. 2022, 20, 310–322. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, Z.; Zhu, C. Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). J. Exp. Bot. 2011, 62, 3563–3573. [Google Scholar] [CrossRef]
- Nejat, N.; Mantri, N. Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit. Rev. Biotechnol. 2018, 38, 93–105. [Google Scholar] [CrossRef]
- Kumar, A.; Gautam, V.; Kumar, P.; Mukherjee, S.; Verma, S.; Sarkar, A.K. Identification and co-evolution pattern of stem cell regulator miR394s and their targets among diverse plant species. BMC Evol. Biol. 2019, 19, 55. [Google Scholar] [CrossRef]
- Pierce, N.W.; Lee, J.E.; Liu, X.; Sweredoski, M.J.; Graham, R.L.; Larimore, E.A.; Rome, M.; Zheng, N.; Clurman, B.E.; Hess, S.; et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 2013, 153, 206–215. [Google Scholar] [CrossRef]
- Song, J.B.; Gao, S.; Sun, D.; Li, H.; Shu, X.X.; Yang, Z.M. miR394 and LCR are involved in Arabidopsis salt and drought stress responses in an abscisic acid-dependent manner. BMC Plant Biol. 2013, 13, 210. [Google Scholar] [CrossRef]
- Huang, S.Q.; Xiang, A.L.; Che, L.L.; Chen, S.; Li, H.; Song, J.B.; Yang, Z.M. A set of miRNAs from Brassica napus in response to sulphate deficiency and cadmium stress. Plant Biotechnol. J. 2010, 8, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Kuang, L.; Yu, J.; Shen, Q.; Fu, L.; Wu, L. Identification of microRNAs responding to aluminium, cadmium and salt stresses in barley roots. Plants 2021, 10, 2754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, H.J.; Qu, D.; Zhu, Z.Z.; Yang, Y.Z.; Zhao, Z.Y. The MdBBX22-miR858-MdMYB9/11/12 module regulates proanthocyanidin biosynthesis in apple peel. Plant Biotechnol. J. 2022, 20, 1683–1700. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Wang, H.; Chi, R.; Qiao, Y.; Wei, J.; Zhang, Y.; Han, M.; Wang, Y.; Li, H. The eTM-miR858-MYB62-like module regulates anthocyanin biosynthesis under low-nitrogen conditions in Malus spectabilis. New Phytol. 2023, 238, 2524–2544. [Google Scholar] [CrossRef]
- Lv, X.; Zhao, Y.; Zhao, Q.; Zhao, L.; Yang, Z.; Wu, Y. The LasmiR858-MYB111 module enhances the antioxidant capacity of green leaf lettuce by promoting flavonoid biosynthesis. Plant Growth Regul. 2024, 103, 647–660. [Google Scholar] [CrossRef]
- Sharma, A.; Badola, P.K.; Bhatia, C.; Sharma, D.; Trivedi, P.K. Primary transcript of miR858 encodes regulatory peptide and controls flavonoid biosynthesis and development in Arabidopsis. Nat. Plants 2020, 6, 1262–1274. [Google Scholar] [CrossRef]
- He, G.; Qin, L.; Tian, W.; Meng, L.; He, T.; Zhao, D. Heavy metal transporters-associated proteins in S. tuberosum: Genome-wide identification, comprehensive gene feature, evolution and expression analysis. Genes 2020, 11, 1269. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Tang, F.; Hajkova, P.; Barton, S.C.; Lao, K.; Surani, M.A. MicroRNA expression profiling of single whole embryonic stem cells. Nucleic Acids Res. 2006, 34, e9. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA Member | Sequence | Length (nt) | Control (TPM) | Cu Stress (TPM) | Log2FC |
|---|---|---|---|---|---|
| htu-miR168a | UCGCUUGGUACAGGUCGGGAA | 21 | 1379 | 1402 | 0.02 |
| htu-miR168b | UCGCUUGGUGCAGGUCGGGAA | 21 | 1385 | 3907 | 1.50 |
| htu-miR394a | UUGGCAUUCUGUCCACCUCC | 20 | 15 | 5 | −1.58 |
| htu-miR397a | UCAUUGAGUGCAGCGUUGAUG | 21 | 112 | 5 | −4.49 |
| htu-miR398a | CGUGUUCUCAGGUCGCCCCUG | 21 | 473 | 11 | −5.43 |
| htu-miR398b | UGUGUUCUCAGGUCGCCCCUG | 21 | 5671 | 147 | −5.27 |
| htu-miR398c | UGUGUUCUCAGGUCACCCCUU | 21 | 5 | 8 | 0.68 |
| htu-miR408a | UGCACUGCCUCUUCCCUGGC | 20 | 45 | 19 | −1.24 |
| htu-miR408b | UGCACUGUCUCUUCCCUGGC | 20 | 6 | 1 | −2.58 |
| htu-miR408c | UGCACUGUCUCUUCCCUGUCU | 21 | 16 | 1 | −4.00 |
| htu-miR858a | UUCGUUGUCUGUUCGACCUUG | 21 | 11,740 | 4480 | −1.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Shao, T.; Dong, W.; Lin, J.; Dai, L.; Ma, Y.; Zhou, Z.; Long, X. Identification and Characterization of Copper-Responsive miRNAs and Their Target Genes in Jerusalem Artichoke. Plants 2025, 14, 955. https://doi.org/10.3390/plants14060955
Chen X, Shao T, Dong W, Lin J, Dai L, Ma Y, Zhou Z, Long X. Identification and Characterization of Copper-Responsive miRNAs and Their Target Genes in Jerusalem Artichoke. Plants. 2025; 14(6):955. https://doi.org/10.3390/plants14060955
Chicago/Turabian StyleChen, Xi, Tianyun Shao, Wenhan Dong, Jiayan Lin, Lixiang Dai, Yilong Ma, Zhaosheng Zhou, and Xiaohua Long. 2025. "Identification and Characterization of Copper-Responsive miRNAs and Their Target Genes in Jerusalem Artichoke" Plants 14, no. 6: 955. https://doi.org/10.3390/plants14060955
APA StyleChen, X., Shao, T., Dong, W., Lin, J., Dai, L., Ma, Y., Zhou, Z., & Long, X. (2025). Identification and Characterization of Copper-Responsive miRNAs and Their Target Genes in Jerusalem Artichoke. Plants, 14(6), 955. https://doi.org/10.3390/plants14060955