
Genetic Diversity Analysis and Core Germplasm Collection Construction of Tartary Buckwheat Based on SSR Markers

 ,
,
Abstract
1. Introduction
2. Results
2.1. SSR Polymorphism and Allelic Diversity
2.2. Genetic Diversity Among Wild, Landrace, and Improved Tartary Buckwheat Accessions
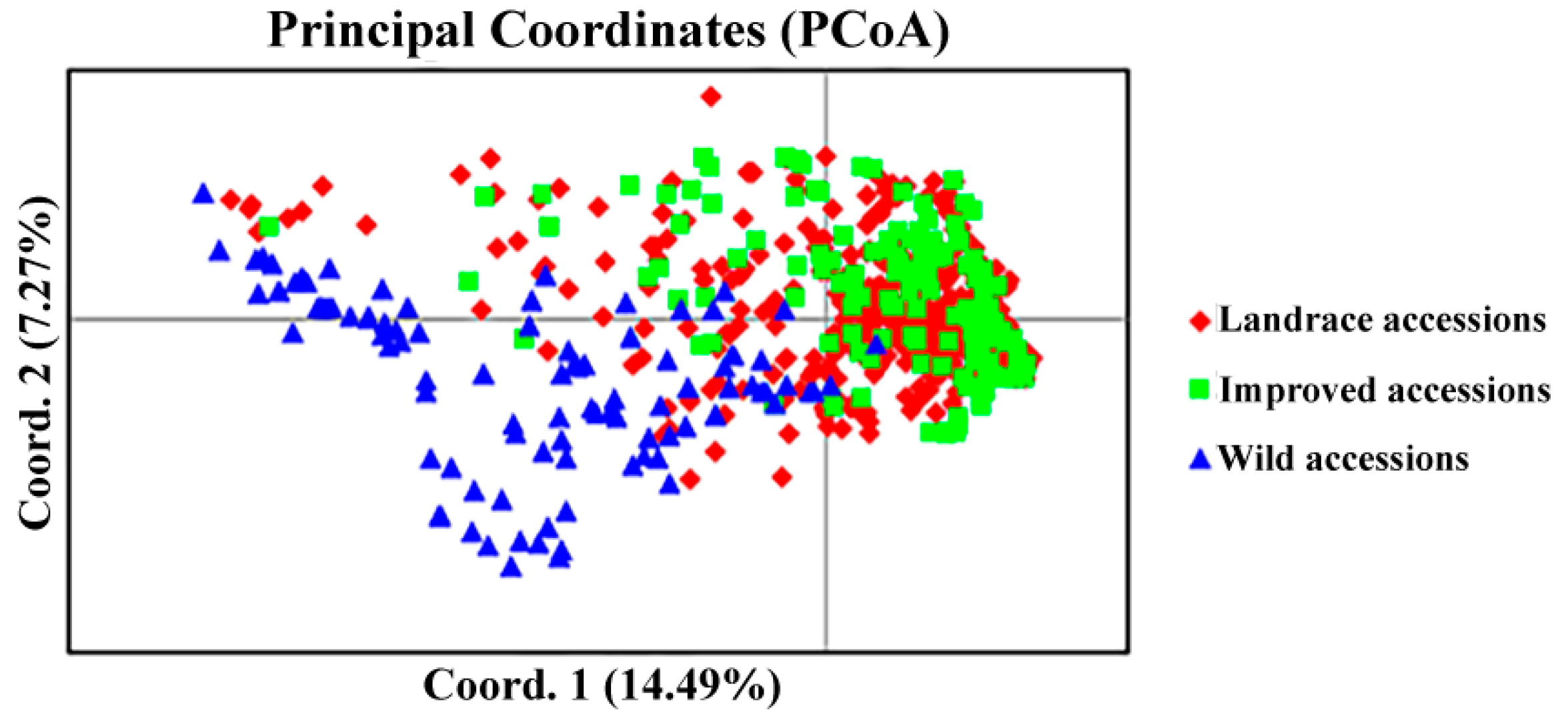
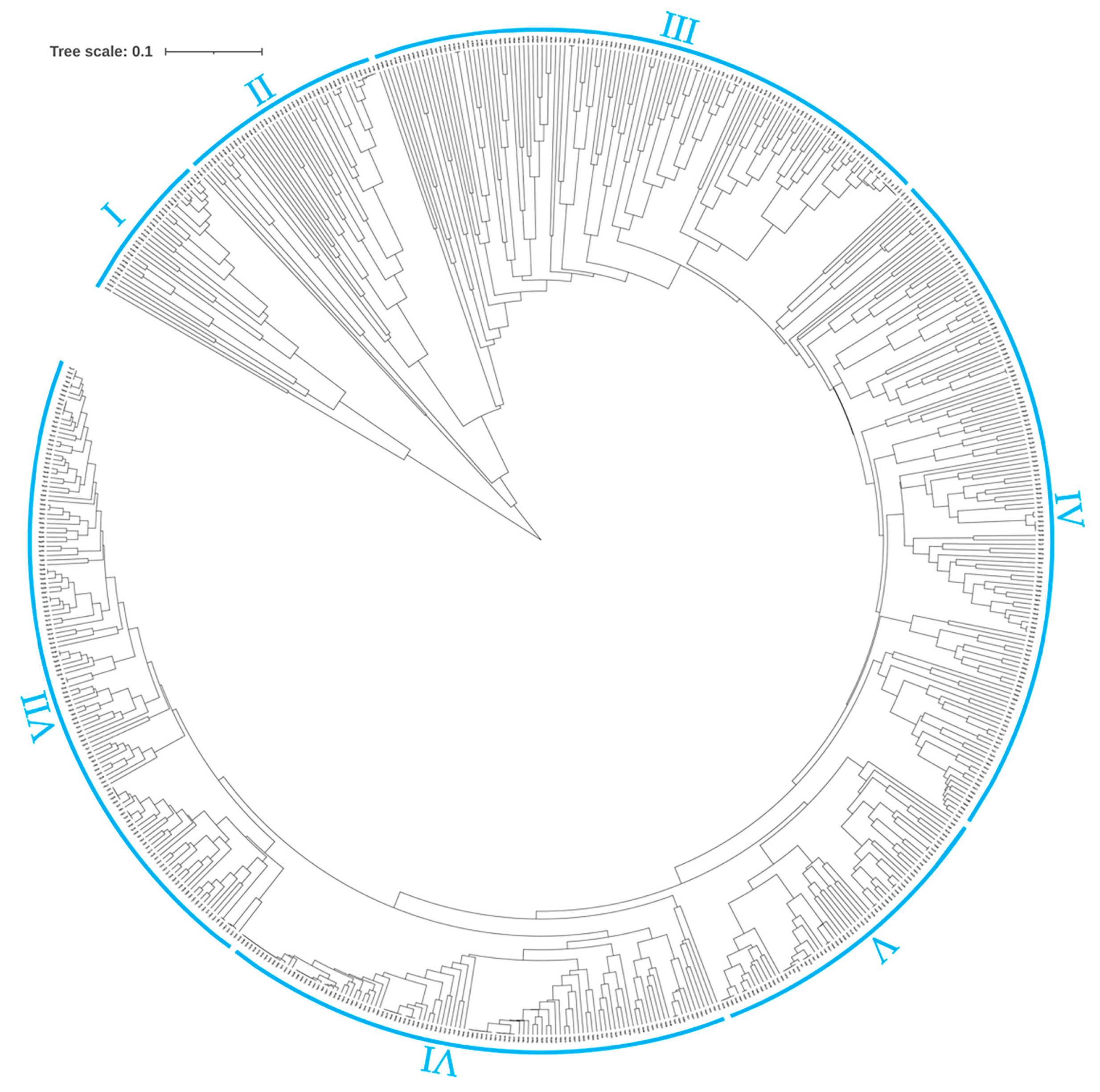
2.3. Genetic Similarity Coefficient Analysis and Cluster Analysis
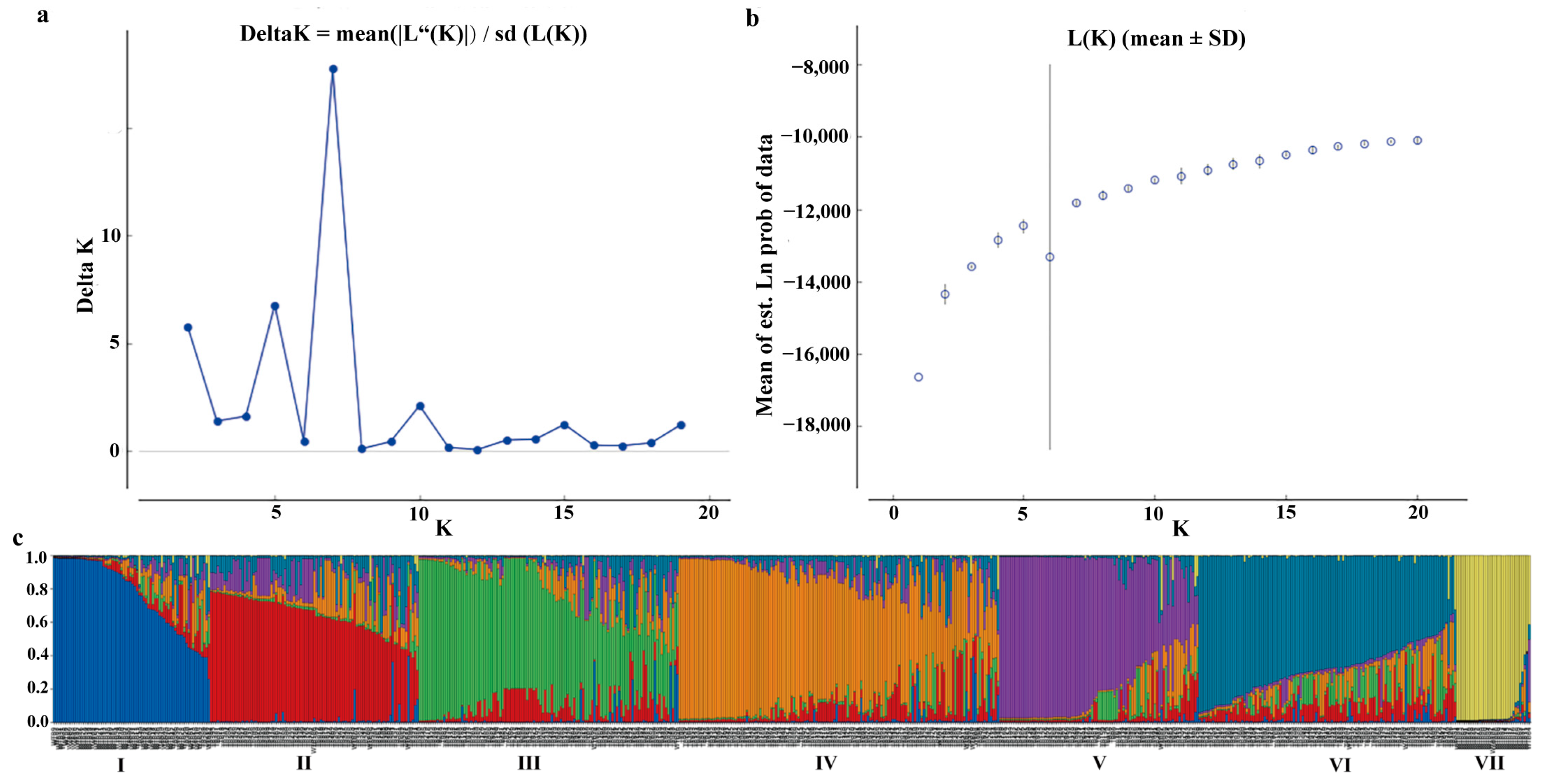
2.4. Analysis of Population Structure
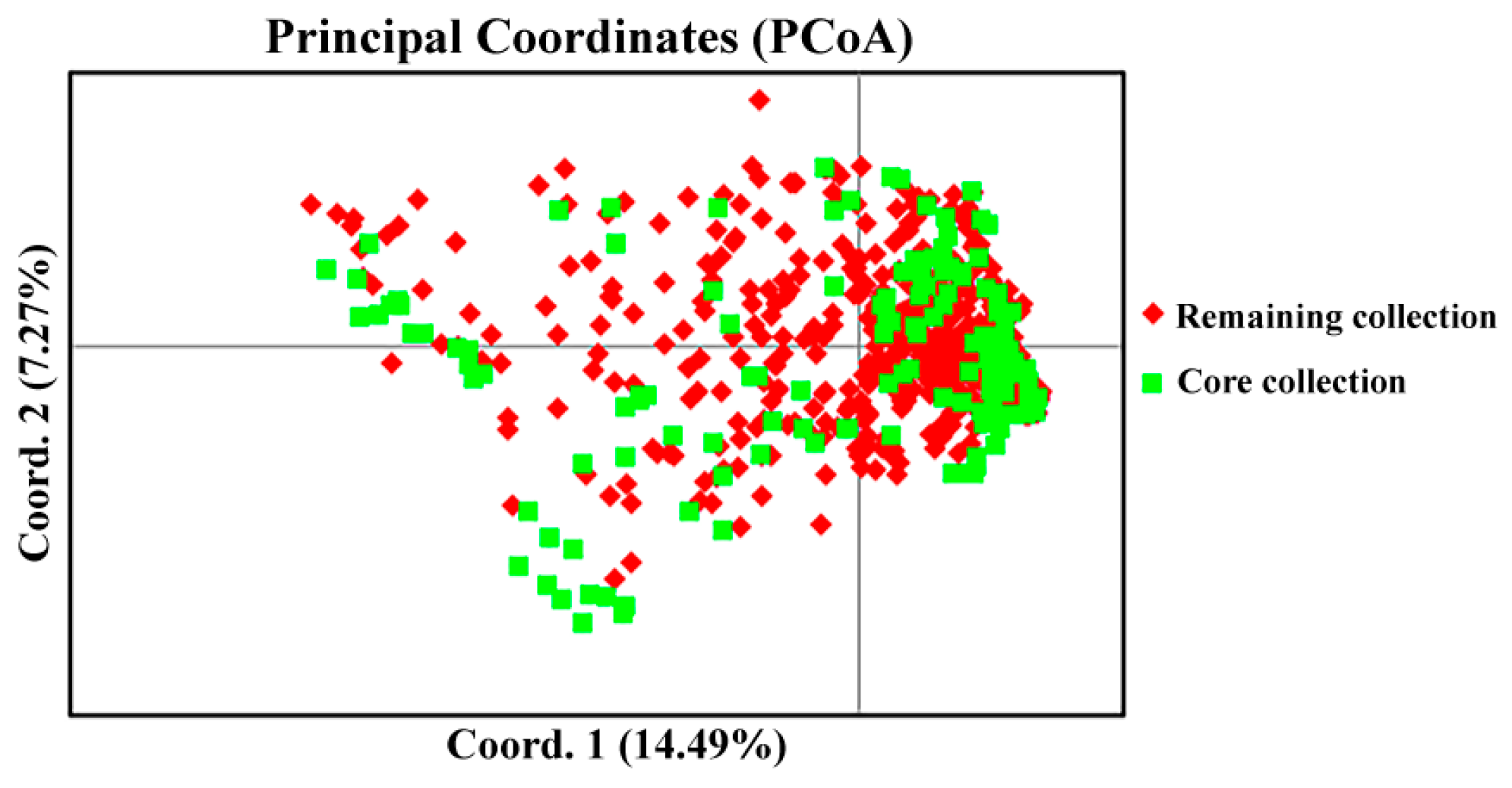
2.5. Construction of Core Collections
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Isolation and PCR Amplification
4.3. Genetic Diversity and Structure Analysis
4.4. Construction and Evaluation of the Core Germplasm Collection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Balážová, Ž.; Čišecká, L.; Gálová, Z.; Hromadová, Z.; Chňapek, M.; Pipan, B.; Meglič, V. Characterization of genetic variability of common and Tartary buckwheat genotypes using microsatellite markers. Plants 2024, 13, 2147. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhuo, C.; Yumei, D.; Dongmei, L.; Keyu, B.; Devra, J.; Jinchao, F.; Chunlin, L. Diversity of Tartary buckwheat (Fagopyrum tataricum) landraces from Liangshan, Southwest China: Evidence from morphology and SSR markers. Agronomy 2022, 12, 1022. [Google Scholar] [CrossRef]
- Zhang, K.; He, M.; Fan, Y.; Zhao, H.; Gao, B.; Yang, K.; Li, F.; Tang, Y.; Gao, Q.; Lin, T.; et al. Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biol. 2021, 22, 23. [Google Scholar] [CrossRef]
- Li, H.; Lv, Q.; Liu, A.; Wang, J.; Sun, X.; Deng, J.; Wu, Q. Comparative metabolomics study of Tartary (Fagopyrum tataricum (L.) Gaertn) and common (Fagopyrum esculentum Moench) buckwheat seeds. Food Chem. 2022, 371, 131125. [Google Scholar] [CrossRef]
- Huda, M.N.; Lu, S.; Jahan, T.; Ding, M.; Jha, R.; Zhang, K.; Zhou, M. Treasure from garden: Bioactive compounds of buckwheat. Food Chem. 2021, 335, 127653. [Google Scholar] [CrossRef]
- Zou, L.; Wu, D.; Ren, G.; Hu, Y.; Peng, L.; Zhao, J.; Xiao, J. Bioactive compounds, health benefits, and industrial applications of Tartary buckwheat (Fagopyrum tataricum). Crit. Rev. Food Sci. Nutr. 2023, 63, 657–673. [Google Scholar] [CrossRef]
- Zhu, Y.; Liang, D.; Song, Z.; Tan, Y.; Guo, X.; Wang, D. Genetic diversity analysis and core germplasm collection construction of Camellia oleifera based on fruit phenotype and SSR data. Genes 2022, 13, 2351. [Google Scholar] [CrossRef]
- Chau, T.P.; Samdani, M.S.; Jhanani, G.K.; Sathiyamoorthi, E.; Lee, J. Metal accumulation and genetic adaptation of Oryza sativa to cadmiun and chromium heavy metal stress: A hydroponic and RAPD analyses. Environ. Res. 2024, 242, 117793. [Google Scholar] [CrossRef]
- Sohn, H.B.; Kim, S.J.; Hong, S.Y.; Park, S.G.; Oh, D.H.; Lee, S.; Nam, H.Y.; Nam, J.H.; Kim, Y.H. Development of 50 InDel-based barcode system for genetic identification of Tartary buckwheat resources. PLoS ONE 2021, 16, e0250786. [Google Scholar] [CrossRef]
- Sun, N.; Chen, J.; Wang, Y.; Hussain, I.; Lei, N.; Ma, X.; Li, W.; Liu, K.; Yu, H.; Zhao, K.; et al. Development and utility of SSR markers based on Brassica sp. whole-genome in triangle of U. Front. Plant Sci. 2024, 14, 1259736. [Google Scholar] [CrossRef]
- Santiago, E.; Caballero, A.; Köpke, C.; Novo, I. Estimation of the contemporary effective population size from SNP data while accounting for mating structure. Mol. Ecol. Resour. 2024, 24, e13890. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, B.; Jiang, D.; Zhuge, F.; Li, Y. Genetic analysis and construction of a fingerprint for licensed Triadica sebifera cultivars using SSR markers. Plants 2024, 13, 1767. [Google Scholar] [CrossRef]
- Agarwal, M.; Shrivastava, N.; Padh, H. Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 2008, 27, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Mallikarjuna, G.; Sameena, S.; Kumar, A.; Gurulakshmi, K.; Reddy, B.V.; Reddy, P.C.O.; Sekhar, A.C. Advancements in molecular marker technologies and their applications in diversity studies. J. Biosci. 2020, 45, 123. [Google Scholar] [CrossRef]
- Kumar, M.; Veena Chaudhary, V.; Sirohi, U.; Sharma, R.V.; Naresh, R.K. Application of molecular markers and their utility in genetic studies of floricultural crops: A review. Int. J. Agric. Biol. Eng. 2019, 12, 229–247. [Google Scholar] [CrossRef]
- Abbasov, M.; Akparov, Z.; Gross, T.; Babayeva, S.; Izzatullayeva, V.; Hajiyev, E.; Rustamov, K.; Gross, P.; Tekin, M.; Akar, T.; et al. Genetic relationship of diploid wheat (Triticum spp.) species assessed by SSR markers. Genet. Resour. Crop Evol. 2018, 65, 1441–1453. [Google Scholar] [CrossRef]
- Nurtaza, A.; Dyussembekova, D.; Shevtsov, A.; Islamova, S.; Samatova, I.; Koblanova, S.; Borodulina, O.; Kakimzhanova, A. Assessing genetic variability and population structure of Alnus glutinosa (black alder) in Kazakhstan using SSR markers. Plants 2024, 13, 3032. [Google Scholar] [CrossRef]
- Misiukevičius, E.; Frercks, B.; Šikšnianienė, J.B.; Kącki, Z.; Gębala, M.; Akulytė, P.; Trilikauskaitė, E.; Stanys, V. Assessing the genetic diversity of daylily germplasm using SSR markers: Implications for daylily breeding. Plants 2023, 12, 1752. [Google Scholar] [CrossRef]
- Sabreena, M.N.; Mahajan, R.; Hashim, M.J.; Iqbal, J.; Alyemeni, M.N.; Bashir, A.G.; Zargar, S.M. Deciphering allelic variability and population structure in buckwheat: An analogy between the efficiency of ISSR and SSR markers. Saudi J. Biol. Sci. 2021, 28, 6050–6056. [Google Scholar] [CrossRef]
- Kishore, G.; Gupta, S.; Pandey, A. Assessment of population genetic diversity of Fagopyrum tataricum using SSR molecular marker. Biochem. Syst. Ecol. 2012, 43, 32–41. [Google Scholar] [CrossRef]
- Pipan, B.; Sinkovič, L.; Neji, M.; Janovská, D.; Zhou, M.; Meglič, V. Agro-morphological and molecular characterization reveal deep insights in promising genetic diversity and marker-trait associations in Fagopyrum esculentum and Fagopyrum tataricum. Plants 2023, 12, 3321. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhou, F.; Gao, Y.; Liu, C.; Yu, S.; Zhao, K.; Gong, W.; Lang, J.; Zhang, H.; Yu, X. Genetic diversity and primary core collection construction of Turnip (Brassica rapa L. ssp. rapifera Matzg) landraces in Tibet revealed via morphological and SSR Markers. Agronomy 2021, 11, 1901. [Google Scholar] [CrossRef]
- Zhou, J.; He, W.; Wang, J.; Liao, X.; Xiang, K.; Ma, M.; Liu, Z.; Li, Y.; Tembrock, L.R.; Wu, Z.; et al. The pan-plastome of tartary buckwheat (Fagopyrum tataricum): Key insights into genetic diversity and the history of lineage divergence. BMC Plant Biol. 2023, 23, 212. [Google Scholar] [CrossRef]
- Hu, G.; Jiang, Q.; Wang, Z.; Li, Z.; Liao, W.; Shen, D.; Zhong, C. Genetic diversity analysis and core collection construction of the Actinidia chinensis complex (kiwifruit) based on SSR markers. Agronomy 2022, 12, 3078. [Google Scholar] [CrossRef]
- Nie, X.H.; Wang, Z.H.; Liu, N.W.; Song, L.; Yan, B.Q.; Xing, Y.; Zhang, Q.; Fang, K.F.; Zhao, Y.L.; Chen, X.; et al. Fingerprinting 146 Chinese chestnut (Castanea mollissima Blume) accessions and selecting a core collection using SSR markers. J. Integr. Agric. 2021, 20, 1277–1286. [Google Scholar] [CrossRef]
- Li, R.; Shi, T.; Chen, Q.; Pan, F.; Chen, Q. Construction of EST-SSR fingerprinting and analysis of genetic diversity of thirty-five registered tartary buckwheat cultivars (Fagopyrum tataricum) in China. Plant Sci. J. 2017, 35, 267–275. [Google Scholar]
- He, Y.; Zhang, K.; Shi, Y.; Lin, H.; Huang, X.; Lu, X.; Wang, Z.; Li, W.; Feng, X.; Shi, T.; et al. Genomic insight into the origin, domestication, dispersal, diversification and human selection of Tartary buckwheat. Genome Biol. 2024, 25, 61. [Google Scholar] [CrossRef]
- Ren, C.Z.; Chen, Q.F.; Li, H.Y.; Wei, L.M.; Guo, L.C.; Wang, C.L.; Li, C.H. Tartary buckwheat germplasm evaluation and genetic breeding research perspective. Acta Bot. Boreal. Occident. Sin. 2023, 43, 1251–1260. [Google Scholar]
- Ren, J.; Yu, X.; Wang, X.; Wang, Y.; Xin, X.; Wang, R.; Zhang, Y.; Liu, M.; Xiang, J. The genetic diversity assessment of broomcorn millet (Panicum miliaceum) and the construction of a mini-core collection. Agronomy 2024, 14, 2226. [Google Scholar] [CrossRef]
- Li, C.; Zhang, L.; Peng, Z.; Li, X.; Liu, Z.; Lu, T.; Kang, X.; Yang, J. Genetic relationship analysis and core collection construction of Eucalyptus grandis from Dongmen improved variety base: The largest eucalypt germplasm resource in China. BMC Plant Biol. 2024, 24, 1240. [Google Scholar] [CrossRef]
- Doyle, J.J. A rapid total DNA preparation procedure for fresh plant tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Leiby, T.S.; He, H.G.; Hulce, D.; Sheng, C.; Liu, J. New Technology Improves Size Calling Of Kilobase DNA Fragments. SoftGenetics LLC 200 Innovation Blvd. Available online: www.softgenetics.com (accessed on 1 January 2025).
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Sun, B.; Li, S.; Pi, Z.; Wu, Z.; Wang, R. Assessment of genetic diversity and population structure of exotic sugar beet (Beta vulgaris L.) varieties using three molecular markers. Plants 2024, 13, 2954. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- De Beukelaer, H.; Smýkal, P.; Davenport, G.F.; Fack, V. Core Hunter II: Fast core subset selection based on multiple genetic diversity measures using Mixed Replica search. BMC Bioinform. 2012, 13, 312. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Marker | Na | Ne | Ho | He | PIC | I | Fis |
|---|---|---|---|---|---|---|---|---|
| 1 | SSR23 | 6 | 1.318 | 0.008 | 0.241 | 0.231 | 0.541 | 0.932 |
| 2 | SSR36 | 9 | 1.546 | 0.071 | 0.353 | 0.319 | 0.693 | 0.817 |
| 3 | Ft1ssR21 | 17 | 3.406 | 0.148 | 0.706 | 0.67 | 1.568 | 0.778 |
| 4 | Ft3ssR15 | 6 | 1.811 | 0.343 | 0.448 | 0.422 | 0.912 | 0.300 |
| 5 | Ft5ssR20 | 13 | 3.182 | 0.234 | 0.686 | 0.651 | 1.565 | 0.633 |
| 6 | P128 | 18 | 1.770 | 0.110 | 0.435 | 0.424 | 1.134 | 0.729 |
| 7 | SSRT26 | 7 | 1.018 | 0.015 | 0.018 | 0.018 | 0.072 | 0.310 |
| 8 | SSRT33 | 4 | 1.900 | 0.691 | 0.474 | 0.377 | 0.727 | −0.414 |
| 9 | SSRT39 | 4 | 1.744 | 0.298 | 0.427 | 0.341 | 0.648 | 0.246 |
| 10 | SSRT40 | 7 | 1.258 | 0.104 | 0.205 | 0.193 | 0.447 | 0.503 |
| 11 | SWUFt169 | 5 | 1.476 | 0.03 | 0.323 | 0.296 | 0.636 | 0.895 |
| 12 | SXAU2177 | 13 | 1.749 | 0.082 | 0.428 | 0.401 | 0.97 | 0.823 |
| 13 | SXAU6249 | 13 | 1.813 | 0.082 | 0.448 | 0.434 | 1.088 | 0.827 |
| 14 | TatG0052 | 12 | 2.431 | 0.160 | 0.589 | 0.559 | 1.336 | 0.713 |
| 15 | TatG0200 | 8 | 2.537 | 0.982 | 0.606 | 0.529 | 1.091 | −0.616 |
| Total | - | 142 | 28.959 | 3.358 | 6.387 | 5.865 | 13.428 | 7.476 |
| Average | - | 9.47 | 1.931 | 0.224 | 0.426 | 0.391 | 0.895 | 0.498 |
| Group | Na | Ne | Ho | uHe | I | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Average | Total | Average | Total | Average | Total | Average | Total | Average | |
| Wild accession | 107 | 7.133 a | 49.852 | 3.324 a | 3.371 | 0.225 a | 8.819 | 0.591 a | 18.520 | 1.23 a |
| Landrace accession | 115 | 7.667 a | 26.218 | 1.748 b | 3.413 | 0.228 a | 5.521 | 0.369 b | 10.966 | 0.731 b |
| Improved accession | 66 | 4.400 b | 24.847 | 1.657 b | 3.234 | 0.216 a | 4.865 | 0.326 b | 9.069 | 0.605 b |
| Accession | Wild Accession | Landrace Accession | Improved Accession |
|---|---|---|---|
| Wild accession | - | 2.187 | 1.843 |
| Landrace accession | 0.103 | - | 33.908 |
| Improved accession | 0.119 | 0.007 | - |
| Accession | Wild Accession | Landrace Accession | Improved Accession |
|---|---|---|---|
| Wild accession | - | 0.256 | 0.281 |
| Landrace accession | 0.774 | - | 0.006 |
| Improved accession | 0.755 | 0.994 | - |
| Population | n | Number of Polymorphic Loci | Percentage of Polymorphic Loci (%) | Na | Ne | Ho | uHe | I |
|---|---|---|---|---|---|---|---|---|
| All collection | 659 | 142 | 100 | 9.467 | 1.931 | 0.224 | 0.426 | 0.895 |
| 30% Pre-core collection | 198 | 135 | 95.07 | 9.000 | 2.443 | 0.288 | 0.527 | 1.123 |
| 25% Pre-core collection | 165 | 131 | 92.25 | 8.733 | 2.561 | 0.297 | 0.541 | 1.158 |
| 20% Pre-core collection | 132 | 131 | 92.25 | 8.733 | 2.689 | 0.298 | 0.559 | 1.205 |
| 15% Pre-core collection | 99 | 130 | 91.55 | 8.677 | 2.775 | 0.305 | 0.570 | 1.233 |
| 10% Pre-core collection | 66 | 122 | 85.92 | 8.133 | 3.140 | 0.311 | 0.602 | 1.307 |
| 8% Pre-core collection | 53 | 116 | 81.69 | 7.733 | 3.334 | 0.324 | 0.610 | 1.324 |
| t1 | 0.296 | −1.666 | −0.674 | −1.456 | −1.351 | |||
| t2 | 0.470 | −1.896 | −0.764 | −1.648 | −1.529 | |||
| t3 | 0.472 | −2.190 * | −0.786 | −1.892 | −1.774 | |||
| t4 | 0.516 | −2.385 * | −0.858 | −2.048 | −1.918 | |||
| t5 | 0.853 | −2.811 ** | −0.920 | −2.431 * | −2.229 * | |||
| t6 | 1.170 | −2.913 ** | −0.622 | −2.525 * | −2.326 * |
| Population | n | Na | Ne | Ho | uHe | I |
|---|---|---|---|---|---|---|
| All collection | 659 (100%) | 9.467 | 1.931 | 0.224 | 0.426 | 0.895 |
| Core collection | 165 (25.04%) | 8.733 | 2.561 | 0.297 | 0.541 | 1.158 |
| Reservation collection | 494 (74.96%) | 7.467 | 1.781 | 0.201 | 0.383 | 0.78 |
| t1 | 0.470 | −1.896 | −0.764 | −1.648 | −1.529 | |
| t2 | 1.307 | 0.635 | 0.226 | 0.642 | 0.788 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Zhang, J.; Liu, Z.; Ran, B.; Deng, J.; Huang, J.; Zhu, L.; Shi, T.; Li, H.; Chen, Q. Genetic Diversity Analysis and Core Germplasm Collection Construction of Tartary Buckwheat Based on SSR Markers. Plants 2025, 14, 771. https://doi.org/10.3390/plants14050771
Cheng Y, Zhang J, Liu Z, Ran B, Deng J, Huang J, Zhu L, Shi T, Li H, Chen Q. Genetic Diversity Analysis and Core Germplasm Collection Construction of Tartary Buckwheat Based on SSR Markers. Plants. 2025; 14(5):771. https://doi.org/10.3390/plants14050771
Chicago/Turabian StyleCheng, Yuanzhi, Jing Zhang, Ziyang Liu, Bin Ran, Jiao Deng, Juan Huang, Liwei Zhu, Taoxiong Shi, Hongyou Li, and Qingfu Chen. 2025. "Genetic Diversity Analysis and Core Germplasm Collection Construction of Tartary Buckwheat Based on SSR Markers" Plants 14, no. 5: 771. https://doi.org/10.3390/plants14050771
APA StyleCheng, Y., Zhang, J., Liu, Z., Ran, B., Deng, J., Huang, J., Zhu, L., Shi, T., Li, H., & Chen, Q. (2025). Genetic Diversity Analysis and Core Germplasm Collection Construction of Tartary Buckwheat Based on SSR Markers. Plants, 14(5), 771. https://doi.org/10.3390/plants14050771