
Unravelling the Chloroplast Genome of the Kazakh Apricot (Prunus armeniaca L.) Through MinION Long-Read Sequencing

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area, DNA Extraction, Purification, and Quantification
2.2. Library Preparation and Sequencing
2.3. Data Processing
2.4. Chloroplast Genome Assembly and Annotation
2.5. Assembly Evaluation
3. Results
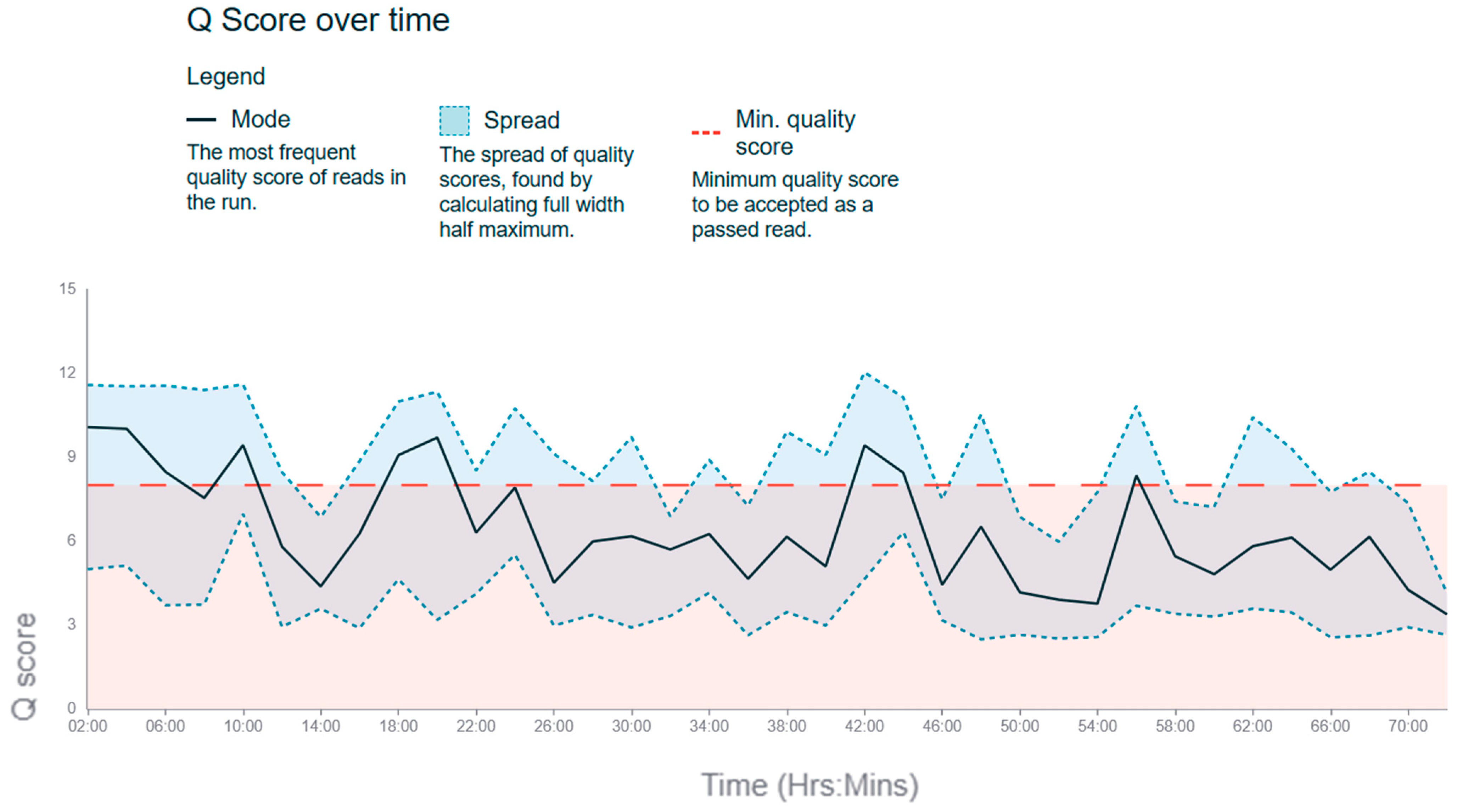
3.1. Assessment of Read Quality
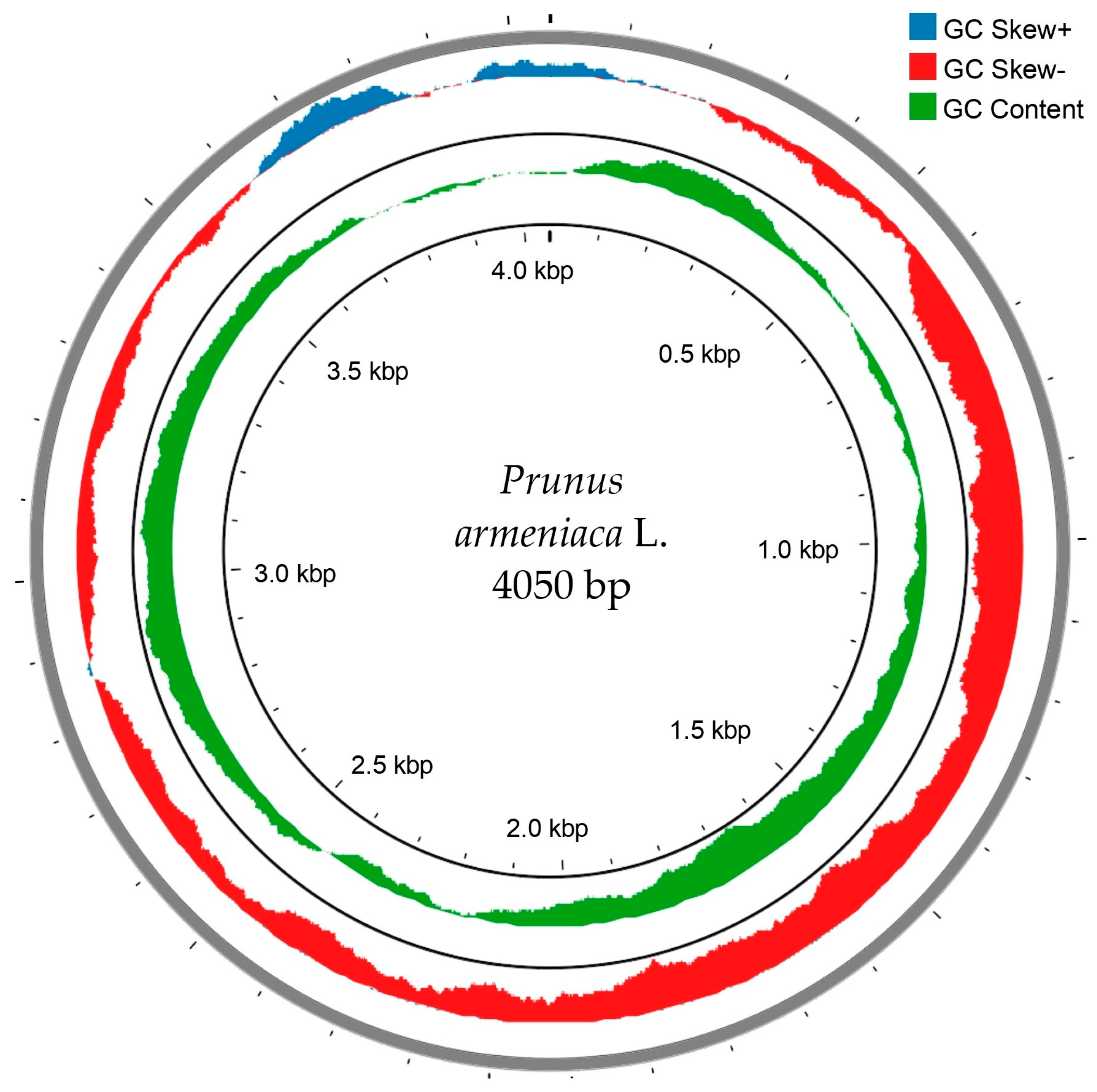
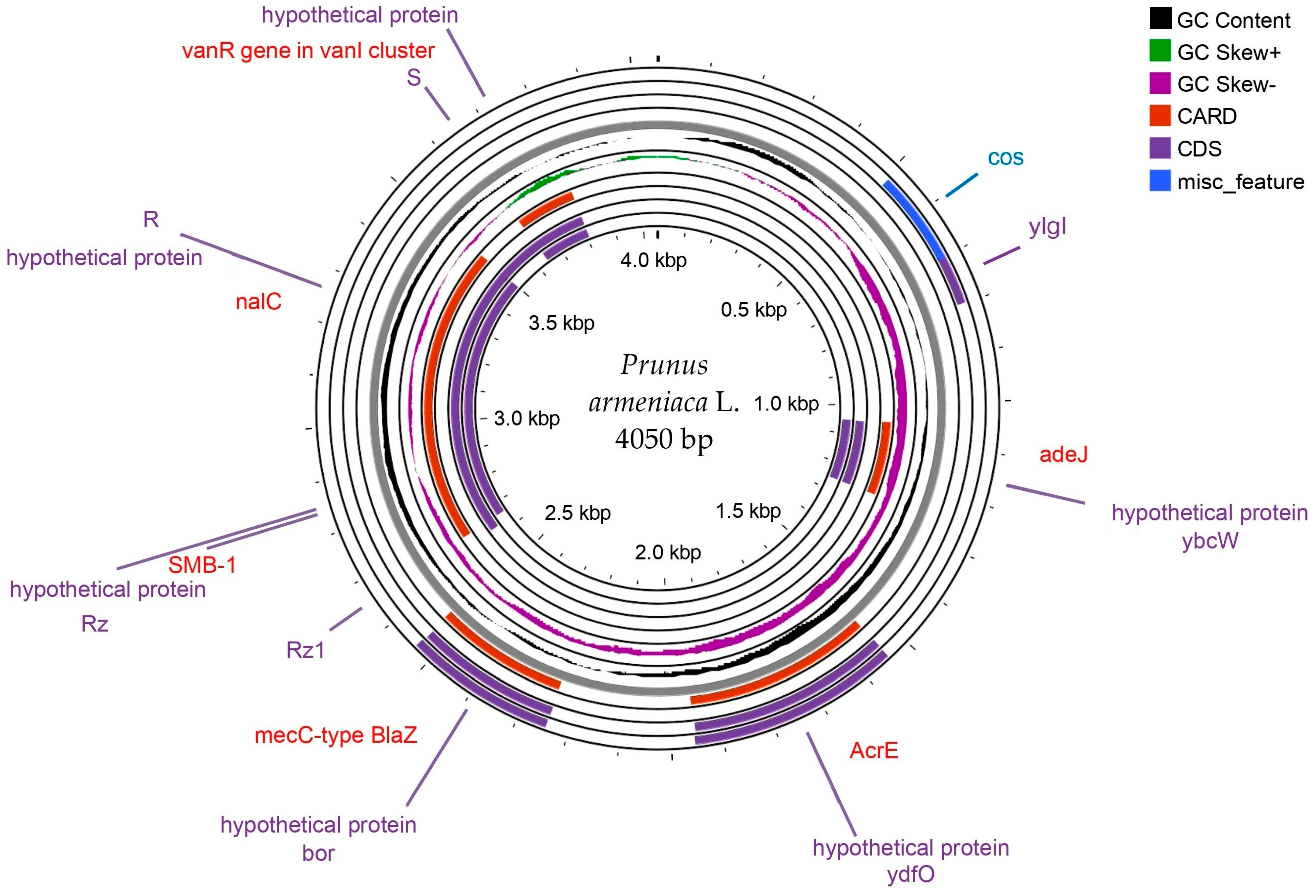
3.2. Chloroplast Genome Assembly
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rakida, A.M. Assessment of genetic diversity of apricot genotypes (Prunus armeniaca L.) from Azerbaijan using SSR markers. Usp. Sovrem. Estestv. Nauk. 2020, 7, 37–42. [Google Scholar]
- Rai, I.; Bachheti, R.K.; Saini, C.K.; Joshi, A.; Satyan, R.S. A review on phytochemical, biological screening, and importance of Wild Apricot (Prunus armeniaca L.). Orient. Pharm. Exp. Med. 2016, 16, 1–15. [Google Scholar] [CrossRef]
- Zaurov, D.E.; Molnar, T.J.; Eisenman, S.W.; Ford, T.M.; Mavlyanova, R.F.; Capik, J.M.; Goffreda, J.C. Genetic resources of apricots (Prunus armeniaca L.) in Central Asia. HortScience 2013, 48, 681–691. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, J.; Wang, S.; Yang, L.; Luo, Y.; Gao, S.; Wang, Y. The apricot (Prunus armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Hortic. Res. 2019, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.A.; Kazantsev, A.V.; Kovalychuk, E.A.; Pavlyukov, M.Y.; Sapkulov, A.V.; Kutaev, D.A.; Borisevich, S.V. Modern hardware and software solutions for whole-genome sequencing, prospects for their implementation in the practice of the troops of radiation, chemical, and biological protection of the Armed Forces of the Russian Federation. Vestn. Voisk RKhB Zashch. 2024, 8, 164–175. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Dreolini, L.; Wilson, J.F.; Balasundaram, M.; Holt, R.A. Complete sequence verification of plasmid DNA using the Oxford Nanopore Technologies’ MinION device. BMC Bioinform. 2023, 24, 116. [Google Scholar] [CrossRef]
- Tytgat, O.; Škevin, S.; Deforce, D.; Van Nieuwerburgh, F. Nanopore sequencing of a forensic combined STR and SNP multiplex. Forensic Sci. Int. Genet. 2022, 56, 102621. [Google Scholar] [CrossRef] [PubMed]
- Fiol, A.; Jurado-Ruiz, F.; López-Girona, E.; Aranzana, M.J. An efficient CRISPR-Cas9 enrichment sequencing strategy for characterising complex and highly duplicated genomic regions: A case study in the Prunus salicina LG3-MYB10 genes cluster. Plant Methods 2022, 18, 105. [Google Scholar] [CrossRef]
- Ryabushkina, N.; Gemedjieva, N.; Kobaisy, M.; Cantrell, C.L. Brief review of Kazakhstan flora and use of its wild species. Asian Australas. J. Plant Sci. Biotechnol. 2008, 2, 64–71. [Google Scholar]
- FAOSTAT. Food and Agriculture Organization of the United Nations; Statistical database; FAO: Rome, Italy, 2013.
- Pucker, B.; Irisarri, I.; de Vries, J.; Xu, B. Plant genome sequence assembly in the era of long reads: Progress, challenges and future directions. Quant. Plant Biol. 2022, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Dumschott, K.; Schmidt, M.H.; Chawla, H.S.; Snowdon, R.; Usadel, B. Oxford Nanopore sequencing: New opportunities for plant genomics? J. Exp. Bot. 2020, 71, 5313–5322. [Google Scholar] [CrossRef]
- Zheng, P.; Zhou, C.; Ding, Y.; Liu, B.; Lu, L.; Zhu, F.; Duan, S. Nanopore sequencing technology and its applications. MedComm 2023, 4, e316. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Chanderbali, A.; Lee, S.; Soltis, D.E.; Soltis, P.S.; Kim, S. High-molecular-weight DNA extraction for long-read sequencing of plant genomes: An optimisation of standard methods. Appl. Plant Sci. 2023, 11, e11528. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, J.; Krawczyk, K.; Paukszto, Ł.; Maździarz, M.; Kurzyński, M.; Szablińska-Piernik, J.; Szczecińska, M. Nanopore Sequencing Technology as an Emerging Tool for Diversity Studies of Plant Organellar Genomes. Diversity 2024, 16, 173. [Google Scholar] [CrossRef]
- Jung, H.; Winefield, C.; Bombarely, A.; Prentis, P.; Waterhouse, P. Tools and strategies for long-read sequencing and de novo assembly of plant genomes. Trends Plant Sci. 2019, 24, 700–724. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Núñez, K.; Rodríguez-Ramos, F.; Leiva-Portilla, D.; Ibáñez, C. Brown biotechnology: A powerful toolbox for resolving current and future challenges in the development of arid lands. SN Appl. Sci. 2020, 2, 1187. [Google Scholar] [CrossRef]
- Imanbayeva, A.; Duisenova, N.; Orazov, A.; Sagyndykova, M.; Belozerov, I.; Tuyakova, A. Study of the Floristic, Morphological, and Genetic (atpF–atpH, Internal Transcribed Spacer (ITS), matK, psbK–psbI, rbcL, and trnH–psbA) Differences in Crataegus ambigua Populations in Mangistau (Kazakhstan). Plants 2024, 13, 1591. [Google Scholar] [CrossRef]
- Imanbayeva, A.; Mukhtubayeva, S.; Adamzhanova, Z.; Duisenova, N.; Zharassova, D.; Lukmanov, A.; Aidyn, O. Phylogenetic analysis of the relict species Dryopteris filix-mas (L.) Schott. by the chloroplast gene (rbcL) and features of modern ontogenesis on the Mangistau Peninsula, Kazakhstan. Casp. J. Environ. Sci. 2024, 22, 849–860. [Google Scholar]
- Qiao, P.; Li, X.; Liu, D.; Lu, S.; Zhi, L.; Rysbekova, A.; Hu, Y.G. Mining novel genomic regions and candidate genes of heading and flowering dates in bread wheat by SNP- and haplotype-based GWAS. Mol. Breed. 2023, 43, 76. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for preparing plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Das, S.; Bandyopadhyay, M.; Bera, S. Optimization of protocol for isolation of genomic DNA from leaves of Selaginella species suitable for RAPD analysis and study of their genetic variation. Am. Fern J. 2012, 102, 47–54. [Google Scholar] [CrossRef]
- Pavelka, J.; Kozelková, T. A new technology for the synthesis of long DNA strings. Int. J. Nanotechnol. Med. Eng. 2017, 2, 127–130. [Google Scholar] [CrossRef]
- Milavetz, B.; Haugen, J.; Rowbotham, K. Mapping the location of nucleosomes in Simian Virus 40 chromatin using the New England Biolabs FS Library Prep Kit. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zeng, Y.; Martin, C.H. Oxford Nanopore sequencing in a research-based undergraduate course. BioRxiv 2017, 227439. [Google Scholar] [CrossRef]
- Jalili, V.; Afgan, E.; Gu, Q.; Clements, D.; Blankenberg, D.; Goecks, J.; Nekrutenko, A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 2020, 48, W395–W402. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network base-calling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Quinlan, A.R. Poretools: A toolkit for analysing nanopore sequence data. Bioinformatics 2014, 30, 3399–3401. [Google Scholar] [CrossRef]
- Kim, C.; Pongpanich, M.; Porntaveetus, T. Unraveling metagenomics through long-read sequencing: A comprehensive review. J. Transl. Med. 2024, 22, 111. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Salzberg, S.L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 2020, 16, e1007981. [Google Scholar]
- Munoz-Barrera, A.; Rubio-Rodriguez, L.A.; Jaspez, D.; Corrales, A.; Marcelino-Rodriguez, I.; Lorenzo-Salazar, J.M.; Flores, C. Benchmarking of bioinformatics tools for the hybrid de novo assembly of human whole-genome sequencing data. bioRxiv 2024. [Google Scholar] [CrossRef]
- Nie, F.; Ni, P.; Huang, N.; Zhang, J.; Wang, Z.; Xiao, C.; Wang, J. De novo diploid genome assembly using long noisy reads. Nat. Commun. 2024, 15, 2964. [Google Scholar] [CrossRef] [PubMed]
- Southwood, D.; Rane, R.V.; Lee, S.F.; Oakeshott, J.G.; Ranganathan, S. Pyro: A comprehensive pipeline for eukaryotic genome assembly. bioRxiv 2023. [Google Scholar] [CrossRef]
- Jayakumar, V.; Nishimura, O.; Kadota, M.; Hirose, N.; Sano, H.; Murakawa, Y.; Sakakibara, Y. Chromosomal-scale de novo genome assemblies of cynomolgus macaque and common marmoset. Sci. Data 2021, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Liu, P.Y.; Shih, P.W. Homopolish: A method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 2021, 22, 95. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long read assembly. Bioinformatics 2019, 35, 2007–2011. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seppey, M.; Manni, M.; Zdobnov, E. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 2019, 227–245. [Google Scholar]
- Vdovina, T.A.; Isakova, E.A.; Lagus, O.A.; Sumbembayev, A.A. Selection assessment of promising forms of natural Hippophae rhamnoides (Elaeagnaceae) populations and their offspring in the Kazakhstan Altai Mountains. Biodiversitas J. Biol. Divers. 2024, 25, 1809–1822. [Google Scholar] [CrossRef]
- Bourguiba, H.; Lasnier, A.; Krška, B.; Zhebentyaeva, T.; Remay, A.; D’Onofrio, C.; Audergon, J.M. Genetic structure of a worldwide germplasm collection of Prunus armeniaca L. In Proceedings of the XVI International Symposium on Apricot Breeding and Culture, Shengyang, China, 29 June–3 July 2015; pp. 203–206. [Google Scholar]
- Fedorenko, O.M.; Zaretskaya, M.V.; Lebedeva, O.N. Genetic and epigenetic mechanisms of plant adaptation (using the model species Arabidopsis thaliana). Tr. Karel. Sci. Cent. Russ. Acad. Sci. 2021, 11, 5–21. [Google Scholar]
- da Câmara Machado, A.; da Câmara Machado, M.L. Genetic transformation in Prunus armeniaca L. (apricot). In Plant Protoplasts and Genetic Engineering VI; Springer: Berlin/Heidelberg, Germany, 1995; pp. 240–254. [Google Scholar]
- Pautov, A.A.; Melnikova, A.N.; Yakovleva, O.V. Two directions of leaf evolution in acacias under arid conditions. Biol. Commun. 2010, 2, 91–99. [Google Scholar]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Orazov, A.; Yermagambetova, M.; Myrzagaliyeva, A.; Mukhitdinov, N.; Tustubayeva, S.; Turuspekov, Y.; Almerekova, S. Plant height variation and genetic diversity between Prunus ledebouriana (Schlecht.) YY Yao and Prunus tenella Batsch using SSR markers in East Kazakhstan. PeerJ 2024, 12, e16735. [Google Scholar] [CrossRef]
- Hernández-Álvarez, C.; García-Oliva, F.; Cruz-Ortega, R.; Romero, M.F.; Barajas, H.R.; Piñero, D.; Alcaraz, L.D. Squash root microbiome transplants and metagenomic inspection for in situ arid adaptations. Sci. Total Environ. 2022, 805, 150136. [Google Scholar] [CrossRef] [PubMed]
- Dyussibayeva, E.; Abylkairova, M.; Tsygankov, V.; Zhirnova, I.; Zeinullina, A.; Yessenbekova, G.; Rysbekova, A. Evaluation of the agronomic traits and correlation analysis of phenotypes of proso millet (Panicum miliaceum L.) germplasm in Kazakhstan. Braz. J. Biol. 2024, 84, e287947. [Google Scholar]
- Rysbekova, A.B.; Kazkeyev, D.T.; Usenbekov, B.N.; Mukhina, Z.M.; Zhanbyrbaev, E.A.; Sartbaeva, I.A.; Zhambakin, K.Z.; Berkimbay, K.A.; Batayeva, D.S. Prebreeding Selection of Rice with Colored Pericarp Based on Genotyping Rc and Pb Genes. Russ. J. Genet. 2017, 53, 43–53. [Google Scholar] [CrossRef]
- Zhirnova, I.; Rysbekova, A.; Dyussibayeva, E.; Zhakenova, A.; Amantaev, B.; Hu, Y.-G.; Feng, B.-L.; Zhunusbayeva, Z. Pre-breeding for waxy proso millet by phenotyping and marker-assisted selection. Chil. J. Agric. Res. 2021, 81, 518–526. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Mean | Median | Minimum Length | Maximum Length | Standard Deviation |
|---|---|---|---|---|---|
| Read length | 1679.33 bp | 1008.5 bp | 63 bp | 31,144 bp | 1622.87 bp |
| GC content | 40.25% | 40.26% | 13.98% | 73.46% | 6.02% |
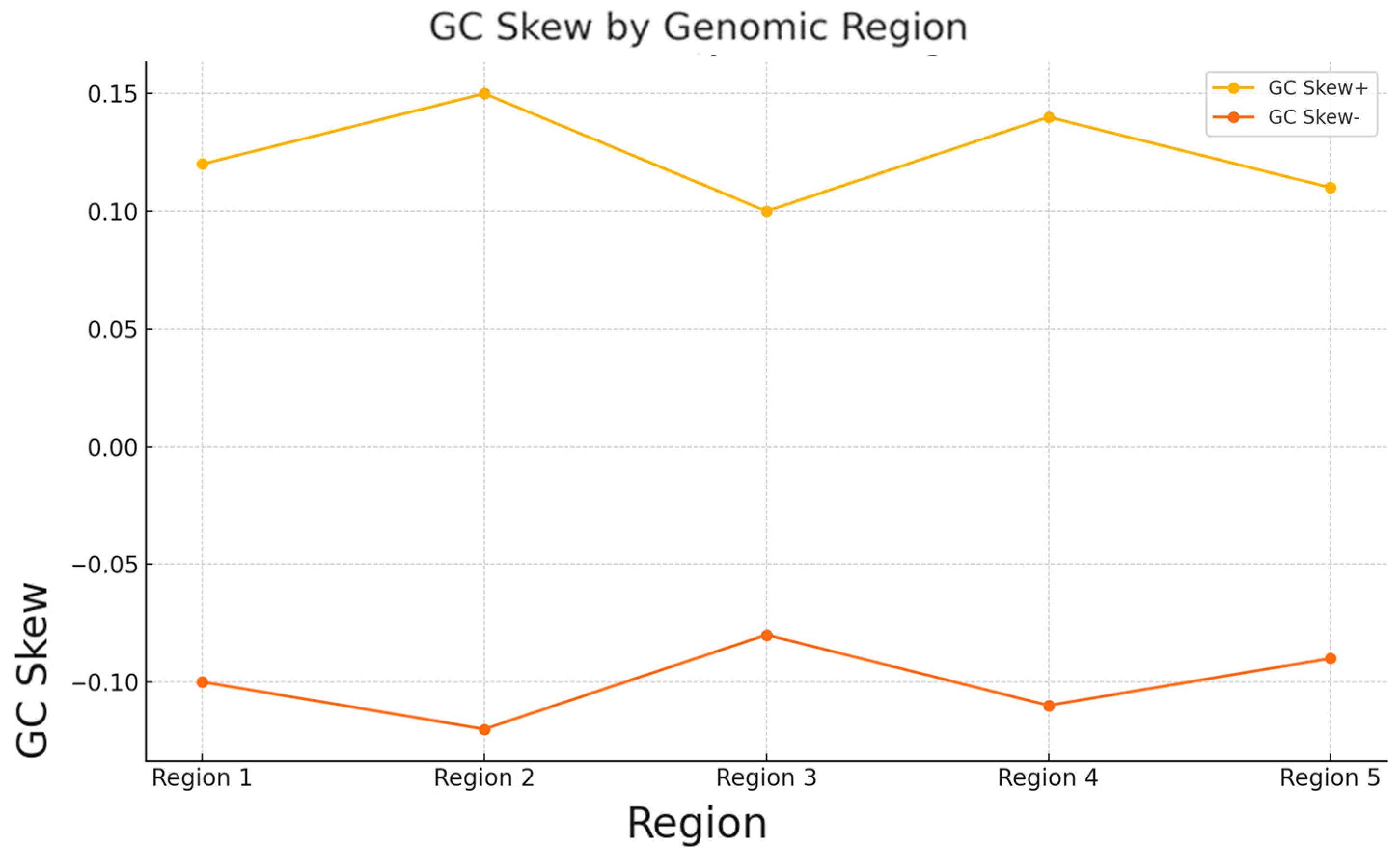
| Region | GC_Content (%) | GC_Skew+ | GC_Skew− | Genes | Protein Function | Adaptation Role |
|---|---|---|---|---|---|---|
| Region 1 | 40.25 | 0.12 | −0.1 | nalC | Regulatory protein for efflux pump | Potentially linked to environmental stress response |
| Region 2 | 42.3 | 0.15 | −0.12 | AcrE | Efflux transporter | Involved in multidrug resistance |
| Region 3 | 39.8 | 0.1 | −0.08 | mecC-type BlaZ | Beta-lactamase is related to antibiotic resistance. | Confers resistance to beta-lactam antibiotics |
| Region 4 | 41.15 | 0.14 | −0.11 | vanR | Regulator in vancomycin cluster | Regulates response to glycopeptide antibiotics |
| Region 5 | 38.9 | 0.11 | −0.09 | hypothetical | Hypothetical protein | Unknown function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akzhunis, I.; Dinara, Z.; Nurzhaugan, D.; Aidyn, O.; Nazerke, T.; Gulmira, T. Unravelling the Chloroplast Genome of the Kazakh Apricot (Prunus armeniaca L.) Through MinION Long-Read Sequencing. Plants 2025, 14, 638. https://doi.org/10.3390/plants14050638
Akzhunis I, Dinara Z, Nurzhaugan D, Aidyn O, Nazerke T, Gulmira T. Unravelling the Chloroplast Genome of the Kazakh Apricot (Prunus armeniaca L.) Through MinION Long-Read Sequencing. Plants. 2025; 14(5):638. https://doi.org/10.3390/plants14050638
Chicago/Turabian StyleAkzhunis, Imanbayeva, Zharassova Dinara, Duisenova Nurzhaugan, Orazov Aidyn, Tolep Nazerke, and Tlepiyeva Gulmira. 2025. "Unravelling the Chloroplast Genome of the Kazakh Apricot (Prunus armeniaca L.) Through MinION Long-Read Sequencing" Plants 14, no. 5: 638. https://doi.org/10.3390/plants14050638
APA StyleAkzhunis, I., Dinara, Z., Nurzhaugan, D., Aidyn, O., Nazerke, T., & Gulmira, T. (2025). Unravelling the Chloroplast Genome of the Kazakh Apricot (Prunus armeniaca L.) Through MinION Long-Read Sequencing. Plants, 14(5), 638. https://doi.org/10.3390/plants14050638