
Transcriptome Analysis of Potato (Solanum tuberosum L.) Seedlings with Varying Resistance Levels Reveals Diverse Molecular Pathways in Early Blight Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Resistance of Virus-Free Potato Seedlings to Early Blight
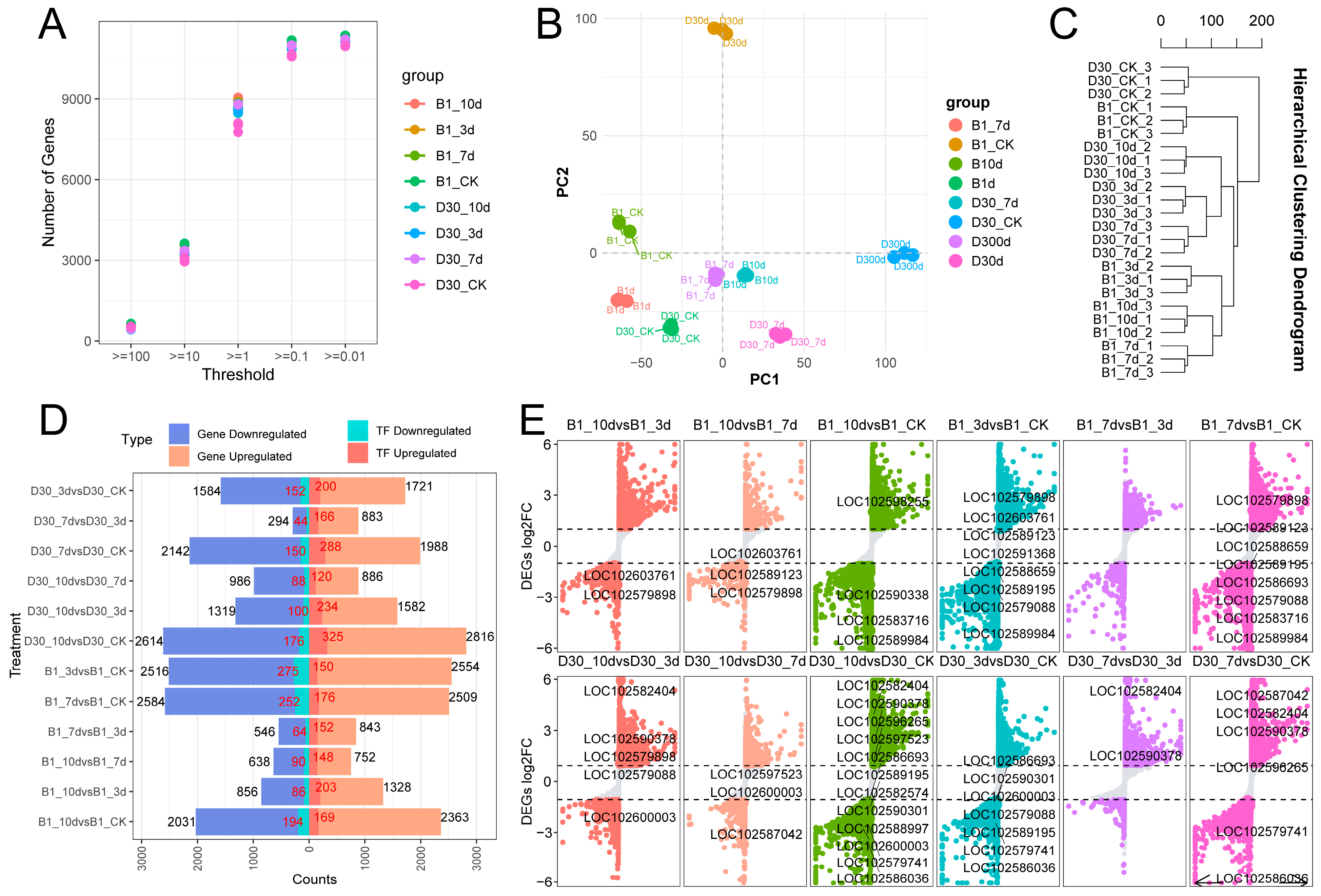
2.2. Details of Transcriptome Sequencing Data
2.3. Differential Expression Gene Analysis
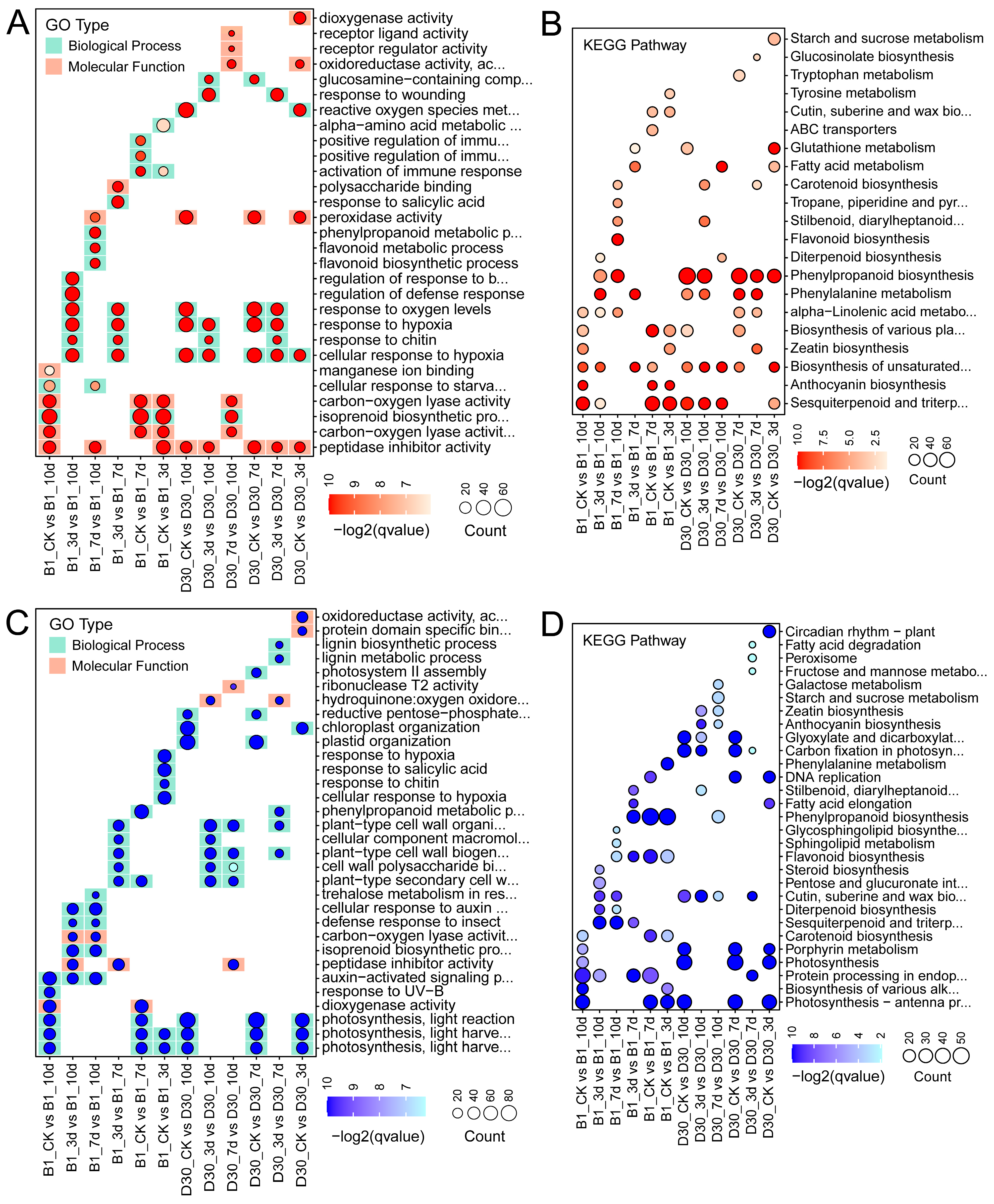
2.4. Functional Enrichment Analysis of Differentially Expressed Genes GO
2.5. Differential Gene Expression Analysis and Trend Cluster Analysis
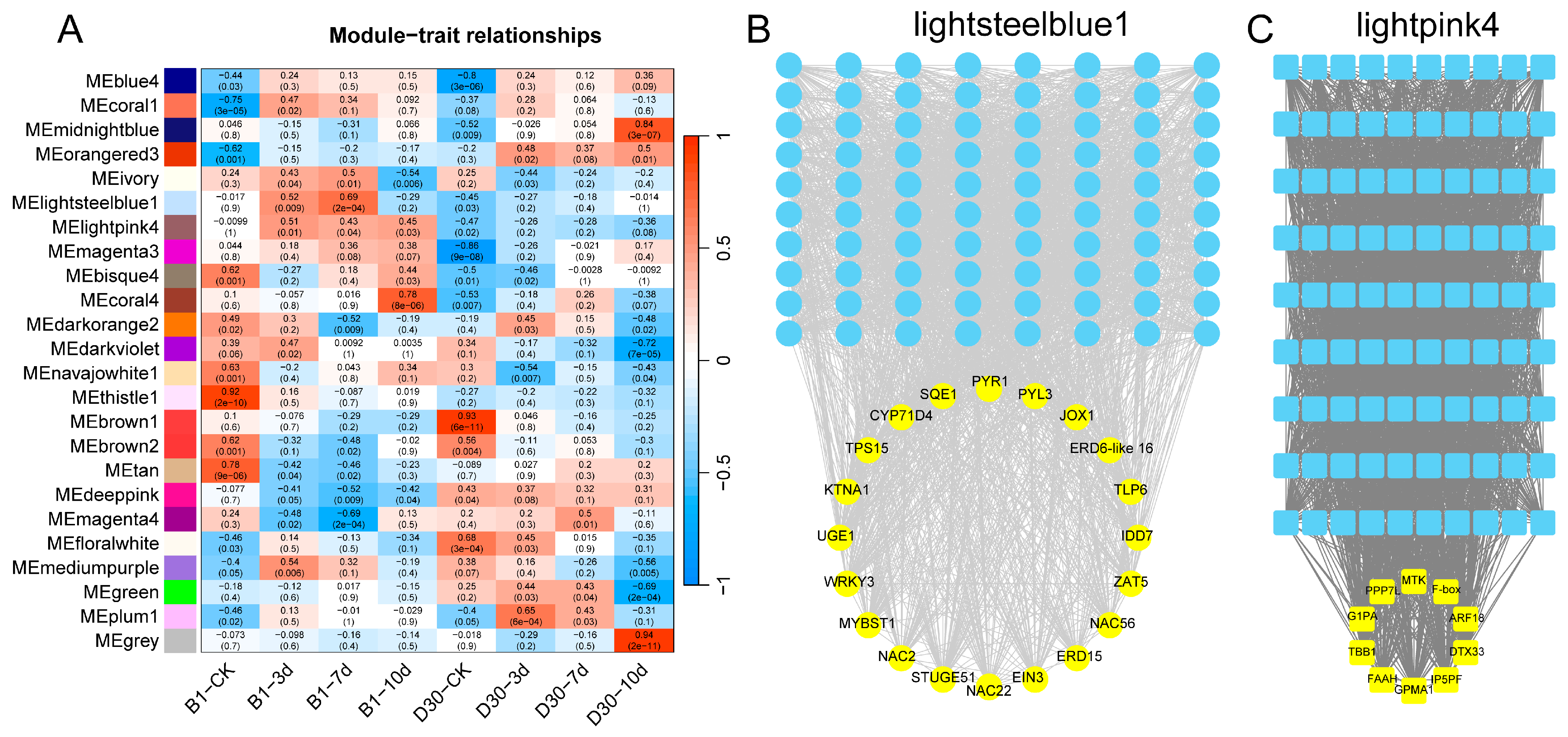
2.6. Regulatory Network Between Genes and TFs Based on WGCNA Analysis
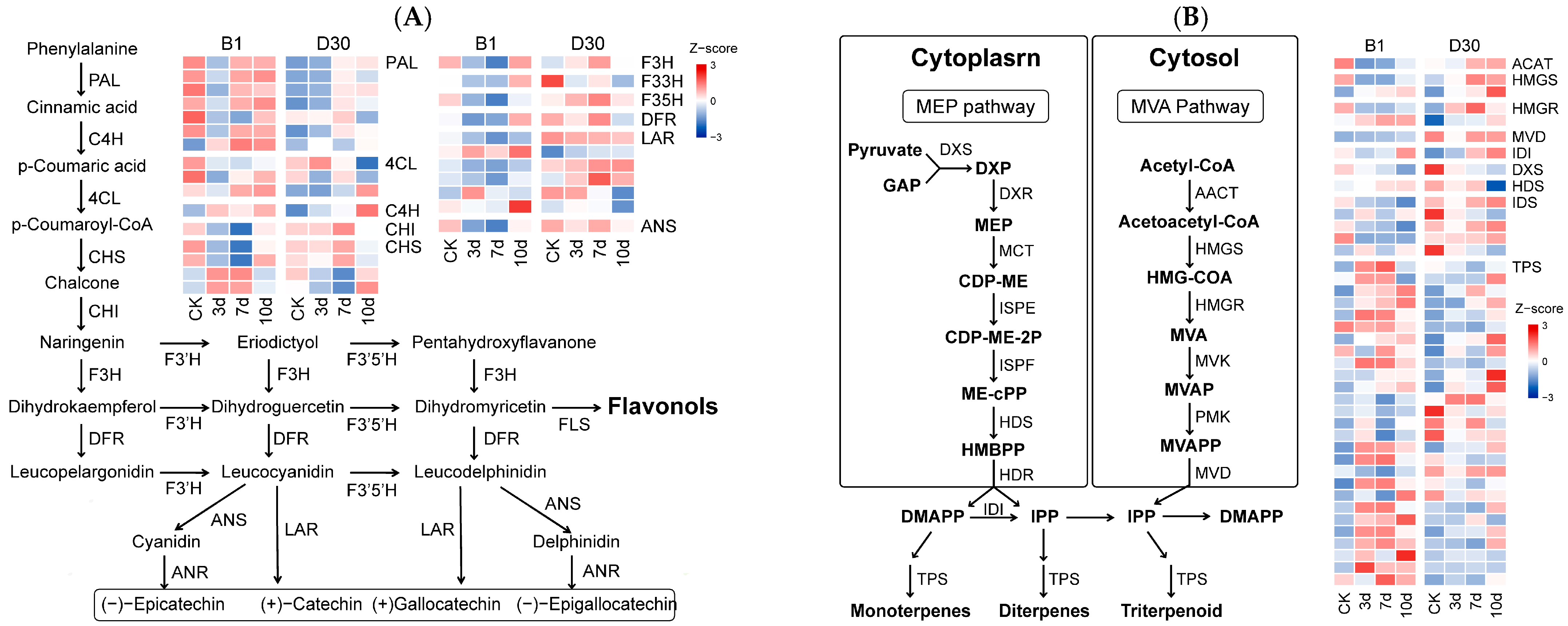
2.7. Key Biosynthesis Pathway Associated with Differential Response to A. solani Infection
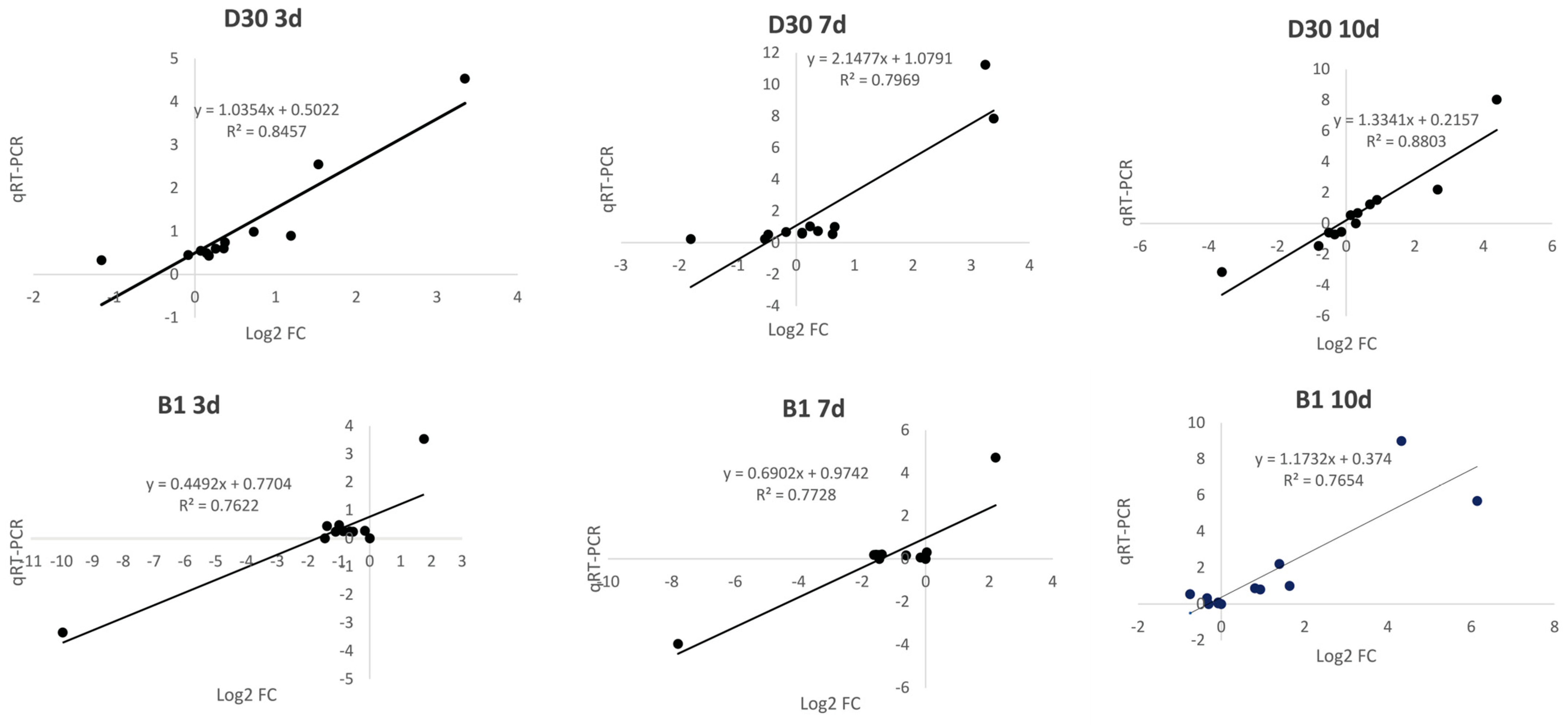
2.8. qRT-PCR Verification of RNA Sequencing Results
3. Discussion
4. Materials and Methods
4.1. Plant Varieties, Test Strains, and Inoculation Method
4.2. RNA Extraction
4.3. Library Construction and High-Throughput Sequencing
4.4. Data Filtering and Gene Annotation
4.5. Differential Gene Analysis and GSEA Enrichment Analysis
4.6. Enrichment Analysis of GO and KEGG Pathways
4.7. Weighted Gene Co-Expression Network Analysis (WGCNA) and Prediction of Hub Gene Network
4.8. Real-Time Fluorescence Quantification
4.9. Data Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Demissie, Y.T. Integrated potato (Solanum tuberosum L.) late blight (Phytophthora infestans) disease management in Ethiopia. Am. J. Biosci. 2019, 7, 123–130. [Google Scholar] [CrossRef]
- Franková, H.; Musilová, J.; Árvay, J.; Harangozo, Ľ.; Šnirc, M.; Vollmannová, A.; Lidiková, J.; Hegedűsová, A.; Jaško, E. Variability of bioactive substances in potatoes (Solanum tuberosum L.) depending on variety and maturity. Agronomy 2022, 12, 1454. [Google Scholar] [CrossRef]
- Wang, R.F.; Su, W.H. The Application of Deep Learning in the Whole Potato Production Chain: A Comprehensive Review. Agriculture 2024, 14, 1225. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Su, W.; Wang, F.; Guo, H. Chapter 18—Potato production in China. In Potato Production Worldwide; Çalişkan, M.E., Bakhsh, A., Jabran, K., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 331–340. [Google Scholar]
- Landschoot, S.; De Reu, J.; Audenaert, K.; Vanhaverbeke, P.; Haesaert, G.; De Baets, B.; Waegeman, W. Potentials and Limitations of Existing Forecasting Models for Alternaria on Potatoes: Challenges for Model Improvement. Potato Res. 2017, 60, 61–76. [Google Scholar] [CrossRef]
- Landschoot, S.; Vandecasteele, M.; Carrette, J.; De Baets, B.; Hofte, M.; Audenaert, K.; Haesaert, G. Assessing the Belgian potato Alternaria population for sensitivity to fungicides with diverse modes of action. Eur. J. Plant Pathol. 2017, 148, 657–672. [Google Scholar] [CrossRef]
- Singh, A.; Gaurav, S.S.; Shukla, G.; Rani, P.; Kumar, B.; Kumar, A. Evaluation of mycogenic silver and zinc oxide nanoparticles as potential control agent against early blight (Alternaria solani) of potato (Solanum tuberosum L.). J. Adv. Sci. Res. 2020, 11, 164–172. [Google Scholar]
- Shelake, R.M.; Wagh, S.G.; Patil, A.M.; Červený, J.; Waghunde, R.R.; Kim, J.Y. Heat stress and plant–biotic interactions: Advances and perspectives. Plants 2024, 13, 2022. [Google Scholar] [CrossRef] [PubMed]
- Adolf, B.; Andrade-Piedra, J.; Bittara Molina, F.; Przetakiewicz, J.; Hausladen, H.; Kromann, P.; Lees, A.; Lindqvist-Kreuze, H.; Perez, W.; Secor, G.A. Fungal, oomycete, and plasmodiophorid diseases of potato. In The Potato Crop: Its Agricultural, Nutritional and Social Contribution to Humankind; Springer International Publishing: Cham, Switzerland, 2020; pp. 307–350. [Google Scholar]
- Abuley, I.K.; Nielsen, B.J.; Hansen, H.H. The influence of crop rotation on the onset of early blight (Alternaria solani). J. Phytopathol. 2019, 167, 35–40. [Google Scholar] [CrossRef]
- Abuley, I.K.; Nielsen, B.J.; Hansen, H.H. The influence of timing the application of nitrogen fertilizer on early blight (Alternaria solani). Pest Manag. Sci. 2019, 75, 1150–1158. [Google Scholar] [CrossRef]
- Brouwer, S.M.; Brus-Szkalej, M.; Saripella, G.V.; Liang, D.; Liljeroth, E.; Grenville-Briggs, L.J. Transcriptome Analysis of Potato Infected with the Necrotrophic Pathogen Alternaria solani. Plants 2021, 10, 2212. [Google Scholar] [CrossRef]
- Pan, C.; Ye, Y.; Ma, Y.; Li, S.; Zhu, Y.; Chu, Y.; Wang, F.; Shen, L.; Tian, J. Cinnamaldehyde triggers fungal apoptosis in Fusarium solani by impairing cell structures and antioxidant defense through restricting glucose availability. Food Biosci. 2025, 69, 106889. [Google Scholar] [CrossRef]
- Pan, X.; Liao, G.; Li, H.; Yuan, W.; Xu, L.; Si, H.; Zhang, J.; Song, J.; Chen, S.; Wang, Z. Citral-based amide derivatives improve the antifungal activity of Kresoxim-methyl against Rhizoctonia solani. Pest Manag. Sci. 2025. [Google Scholar] [CrossRef]
- Dong, S.; Zhou, S. Potato late blight caused by Phytophthora infestans: From molecular interactions to integrated management strategies. J. Integr. Agric. 2022, 21, 3456–3466. [Google Scholar] [CrossRef]
- Bourke, P.A. Emergence of Potato blight, 1843–46. CABI Digit. Libr. 1964, 203, 805–808. [Google Scholar]
- Lafferty, K.D. The ecology of climate change and infectious diseases. Ecology 2009, 90, 888–900. [Google Scholar] [CrossRef]
- Hooker, W.J. Compendium of Potato Diseases; International Potato Center: Lima, Peru, 1981. [Google Scholar]
- Boyd, A. Potato storage diseases. Rev. Pl. Plath. 1972, 51, 297–321. [Google Scholar]
- Noh, J.S.; Hwang, S.H.; Maung, C.E.H.; Cho, J.-Y.; Kim, K.Y. Enhanced control efficacy of Bacillus subtilis NM4 via integration of chlorothalonil on potato early blight caused by Alternaria solani. Microb. Pathog. 2024, 190, 106604. [Google Scholar] [CrossRef]
- Fu, J.; Ma, B.; Zhu, L.; Zhang, Y.; Li, Y.; Tian, S.; Wang, L.; Liu, H.; Wei, G.; Yu, L. Transcriptomic analysis reveals distinct strategies of two potato cultivars against Phytophthora infestans and identifies StWRKY26 as a positive regulator of late blight resistance. Environ. Exp. Bot. 2025, 229, 106084. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, J.; Bai, Z.; Jiang, H.; Zhu, J.; Yang, Z. Alternaria solani Effector AsCEP20, Essential for Virulence, Targets Potato StFtsH4 Protein to Suppress Plant Disease Resistance. Mol. Plant Pathol. 2025, 26, e70109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yu, S.; Yang, Y.; Zhang, J.; Zhao, D.; Pan, Y.; Fan, S.; Yang, Z.; Zhu, J. Antifungal effects of volatiles produced by Bacillus subtilis against Alternaria solani in potato. Front. Microbiol. 2020, 11, 1196. [Google Scholar] [CrossRef] [PubMed]
- Nehela, Y.; Taha, N.; Elzaawely, A.; Xuan, T.; Amin, M.; Ahmed, M.; El-Nagar, A. Benzoic acid and its hydroxylated derivatives suppress early blight of tomato (Alternaria solani) via the induction of salicylic acid biosynthesis and enzymatic and nonenzymatic antioxidant defense machinery. J. Fungi 2021, 7, 663. [Google Scholar] [CrossRef] [PubMed]
- Beliaev, D.; Yuorieva, N.; Tereshonok, D.; Tashlieva, I.; Derevyagina, M.; Meleshin, A.; Rogozhin, E.; Kozlov, S. High resistance of potato to early blight is achieved by expression of the Pro–SmAMP1 gene for hevein-like antimicrobial peptides from common chickweed (Stellaria media). Plants 2021, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Dey, U. An overview of fungal and bacterial biopesticides to control plant pathogens/diseases. Afr. J. Microbiol. Res. 2014, 8, 1749–1762. [Google Scholar] [CrossRef]
- Shahid, M.; Zaidi, A.; Khan, M.S.; Rizvi, A.; Saif, S.; Ahmed, B. Recent advances in management strategies of vegetable diseases. Microb. Strateg. Veg. Prod. 2017, 197–226. [Google Scholar]
- Rafiq, M.; Shoaib, A.; Javaid, A.; Parveen, S.; Hassan, M.A.; Nawaz, H.H.; Cheng, C. Application of Asteraceae biomass and biofertilizers to improve potato crop health by controlling black scurf disease. Front. Plant Sci. 2024, 15, 1437702. [Google Scholar] [CrossRef]
- Rafiq, M.; Javaid, A.; Kanwal, A.; Anwar, A.; Khan, I.H.; Kanwal, Q.; Cheng, C. GC-MS analysis and antifungal potential of flower extract of Acacia nilotica subsp. Indica against Macrophomina phaseolina. Microb. Pathog. 2024, 194, 106819. [Google Scholar] [CrossRef]
- Murmu, S.; Dey, S.; Chakraborty, A. Management of early blight of potato using bio control agents and plant extracts. J. Appl. Nat. Sci. 2015, 7, 860–864. [Google Scholar] [CrossRef]
- Kumar, S.; Chandra, R. Integrated field application of T. viride, botanicals, and fungicides for managing early blight (Alternaria solani) and enhancement of plant growth, tuber nutritional quality, and potato yield. J. Nat. Pestic. Res. 2024, 8, 100070. [Google Scholar] [CrossRef]
- Xue, W.; Haynes, K.G.; Qu, X. Characterization of Early Blight Resistance in Potato Cultivars. Plant Dis. 2019, 103, 629–637. [Google Scholar] [CrossRef]
- Sajeevan, R.S.; Abdelmeguid, I.; Saripella, G.V.; Lenman, M.; Alexandersson, E. Comprehensive transcriptome analysis of different potato cultivars provides insight into early blight disease caused by Alternaria solani. BMC Plant Biol. 2023, 23, 130. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, S.M.; Odilbekov, F.; Burra, D.D.; Lenman, M.; Hedley, P.E.; Grenville-Briggs, L.; Alexandersson, E.; Liljeroth, E.; Andreasson, E. Intact salicylic acid signalling is required for potato defence against the necrotrophic fungus Alternaria solani. Plant Mol. Biol. 2020, 104, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Odilbekov, F.; Carlson-Nilsson, U.; Liljeroth, E. Phenotyping early blight resistance in potato cultivars and breeding clones. Euphytica 2014, 197, 87–97. [Google Scholar] [CrossRef]
- Dita, M.A.; Brommonschenkel, S.H.; Matsuoka, K.; Mizubuti, E.S.G. Histopathological Study of the Alternaria solani Infection Process in Potato Cultivars with Different Levels of Early Blight Resistance. J. Phytopathol. 2007, 155, 462–469. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.D.; Stevenson, W.R.; Macguidwin, A.E.; Kelling, K.A.; Binning, L.K.; Zhu, J. Plant pathogen population dynamics in potato fields. J. Nematol. 2002, 34, 189–193. [Google Scholar]
- Pasche, J.S.; Wharam, C.M.; Gudmestad, N.C. Shift in sensitivity of Alternaria solani in response to fungicides. Plant Dis. 2004, 88, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Wang, Z.; Guo, Y.; Zhang, X. Comparative transcriptome profiling reveals the role of phytohormones and phenylpropanoid pathway in early-stage resistance against powdery mildew in watermelon (Citrullus lanatus L.). Front. Plant Sci. 2022, 13, 1016822. [Google Scholar] [CrossRef]
- Wei, H.; Yang, M.; Ke, Y.; Liu, J.; Chen, Z.; Zhao, J.; Zhao, Y.; Huang, F.; Yu, L. Comparative physiological and transcriptomic profiles reveal regulatory mechanisms of soft rot disease resistance in Amorphophallus spp. Physiol. Mol. Plant Pathol. 2022, 118, 101807. [Google Scholar] [CrossRef]
- He, Y.; Lan, L.; Zhao, L.; Cai, B.; Qu, S.; Wang, S. Comparative transcriptomics analysis reveals MdGRAS53 contributes to disease resistance against Alternaria blotch of apple. J. Plant Physiol. 2022, 273, 153697. [Google Scholar] [CrossRef]
- Meng, H.; Sun, M.; Jiang, Z.; Liu, Y.; Sun, Y.; Liu, D.; Jiang, C.; Ren, M.; Yuan, G.; Yu, W.; et al. Comparative transcriptome analysis reveals resistant and susceptible genes in tobacco cultivars in response to infection by Phytophthora nicotianae. Sci. Rep. 2021, 11, 809. [Google Scholar] [CrossRef]
- Aguilera-Galvez, C.; Champouret, N.; Rietman, H.; Lin, X.; Wouters, D.; Chu, Z.; Jones, J.D.; Vossen, J.H.; Visser, R.G.; Wolters, P.J.; et al. Two different R gene loci co-evolved with Avr2 of Phytophthora infestans and confer distinct resistance specificities in potato. Stud. Mycol. 2018, 89, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, H.; Aminian, H.; Sahebani, N.; Halterman, D. Effect of Alternaria solani Exudates on Resistant and Susceptible Potato Cultivars from Two Different pathogen isolates. Plant Pathol. J. 2011, 27, 14–19. [Google Scholar] [CrossRef]
- Balaji, V.; Mayrose, M.; Sherf, O.; Jacob-Hirsch, J.; Eichenlaub, R.; Iraki, N.; Manulis-Sasson, S.; Rechavi, G.; Barash, I.; Sessa, G. Tomato transcriptional changes in response to Clavibacter michiganensis subsp. michiganensis reveal a role for ethylene in disease development. Plant Physiol. 2008, 146, 1797–1809. [Google Scholar] [CrossRef]
- Bentham, A.R.; De la Concepcion, J.C.; Mukhi, N.; Zdrzałek, R.; Draeger, M.; Gorenkin, D.; Hughes, R.K.; Banfield, M.J. A molecular roadmap to the plant immune system. J. Biol. Chem. 2020, 295, 14916–14935. [Google Scholar] [CrossRef]
- Kumar, A.; Changwal, C.; Hada, A.; Singh, P.K. Molecular insight of plant–pathogen interaction. In Sustainable Agriculture in the Era of Climate Change; Springer: Berlin/Heidelberg, Germany, 2020; pp. 481–511. [Google Scholar]
- Kunjeti, S.G.; Evans, T.A.; Marsh, A.G.; Gregory, N.F.; Kunjeti, S.; Meyers, B.C.; Kalavacharla, V.S.; Donofrio, N.M. RNA-Seq reveals infection-related global gene changes in Phytophthora phaseoli, the causal agent of lima bean downy mildew. Mol. Plant Pathol. 2012, 13, 454–466. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, J.; Davis, R.E.; Wei, H.; Zong, X.; Wei, W.; Shao, J.; Zhao, Y.; Liu, Q. Transcriptome analysis reveals a complex array of differentially expressed genes accompanying a source-to-sink change in phytoplasma-infected sweet cherry leaves. Ann. Appl. Biol. 2019, 175, 69–82. [Google Scholar] [CrossRef]
- Bian, X.; Zhao, Y.; Xiao, S.; Yang, H.; Han, Y.; Zhang, L. Metabolome and transcriptome analysis reveals the molecular profiles underlying the ginseng response to rusty root symptoms. BMC Plant Biol. 2021, 21, 215. [Google Scholar] [CrossRef]
- Macharia, T.N.; Bellieny-Rabelo, D.; Moleleki, L.N. Transcriptome Profiling of Potato (Solanum tuberosum L.) Responses to Root-Knot Nematode (Meloidogyne javanica) Infestation during a Compatible Interaction. Microorganisms 2020, 8, 1443. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Liu, J.; Pang, J.; Kondo, H.; Chi, S.; Zhang, J.; Sun, L.; Andika, I.B. Common but Nonpersistent Acquisitions of Plant Viruses by Plant-Associated Fungi. Viruses 2022, 14, 2279. [Google Scholar] [CrossRef]
- Yao, M.; Wang, X.; Long, J.; Bai, S.; Cui, Y.; Wang, Z.; Liu, C.; Liu, F.; Wang, Z.; Li, Q. Identification of Key Modules and Candidate Genes for Powdery Mildew Resistance of Wheat-Agropyron cristatum Translocation Line WAT-2020-17-6 by WGCNA. Plants 2023, 12, 335. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Baizan, N.; Ribas, L.; Piferrer, F. Improved biomarker discovery through a plot twist in transcriptomic data analysis. BMC Biol. 2022, 20, 208. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, L.; Ye, H.; Tu, W. Weighted gene co-expression network analysis in biomedicine research. Chin. J. Biotechnol. 2017, 33, 1791–1801. [Google Scholar]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted Gene Coexpression Network Analysis: State of the Art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef]
- Cui, M.; Han, S.; Wang, D.; Haider, M.S.; Guo, J.; Zhao, Q.; Du, P.; Sun, Z.; Qi, F.; Zheng, Z.; et al. Gene Co-expression Network Analysis of the Comparative Transcriptome Identifies Hub Genes Associated with Resistance to Aspergillus flavus L. in Cultivated Peanut (Arachis hypogae L.). Front. Plant Sci. 2022, 13, 899177. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J. Multiple functions and regulatory networks of WRKY33 and its orthologs. Gene 2024, 931, 148899. [Google Scholar] [CrossRef] [PubMed]
- Merz, P.R.; Moser, T.; Höll, J.; Kortekamp, A.; Buchholz, G.; Zyprian, E.; Bogs, J. The transcription factor VvWRKY33 is involved in the regulation of grapevine (Vitis vinifera) defense against the oomycete pathogen Plasmopara viticola. Physiol. Plant 2015, 153, 365–380. [Google Scholar] [CrossRef]
- Ji, Y.; Mou, M.; Zhang, H.; Wang, R.; Wu, S.; Jing, Y.; Zhang, H.; Li, L.; Li, Z.; Chen, L. GhWRKY33 negatively regulates jasmonate-mediated plant defense to Verticillium dahliae. Plant Divers. 2023, 45, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, Y.; Xiao, D.; Li, Y.; Liu, T.; Hou, X. BcWRKY33A Enhances Resistance to Botrytis cinerea via Activating BcMYB51-3 in Non-Heading Chinese Cabbage. Int. J. Mol. Sci. 2022, 23, 8222. [Google Scholar] [CrossRef]
- Wang, R.; Wang, G.-L.; Ning, Y. PALs: Emerging Key Players in Broad-Spectrum Disease Resistance. Trends Plant Sci. 2019, 24, 785–787. [Google Scholar] [CrossRef]
- Bai, Y.; Feng, Z.; Paerhati, M.; Wang, J. Phenylpropanoid metabolism enzyme activities and gene expression in postharvest melons inoculated with Alternaria alternata. Appl. Biol. Chem. 2021, 64, 83. [Google Scholar] [CrossRef]
- Chen, X.; Fang, X.; Zhang, Y.; Wang, X.; Zhang, C.; Yan, X.; Zhao, Y.; Wu, J.; Xu, P.; Zhang, S. Overexpression of a soybean 4-coumaric acid: Coenzyme A ligase (GmPI4L) enhances resistance to Phytophthora sojae in soybean. Funct. Plant Biol. 2019, 46, 304–313. [Google Scholar] [CrossRef]
- Ma, J.; Zuo, D.; Zhang, X.; Li, H.; Ye, H.; Zhang, N.; Li, M.; Dang, M.; Geng, F.; Zhou, H.; et al. Genome-wide identification analysis of the 4-Coumarate: CoA ligase (4CL) gene family expression profiles in Juglans regia and its wild relatives J. Mandshurica resistance and salt stress. BMC Plant Biol. 2024, 24, 211. [Google Scholar] [CrossRef]
- Brolund, A.; Sundqvist, M.; Kahlmeter, G.; Grape, M. Molecular Characterisation of Trimethoprim Resistance in Escherichia coli and Klebsiella pneumoniae during a Two Year Intervention on Trimethoprim Use. PLoS ONE 2010, 5, e9233. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-D.; Wang, S.-P. Analysis and mapping of homologous sequences of barley disease resistance gene Mlo and maize disease resistance gene Hm1 in rice. Acta Genet. Sin. 2002, 29, 875–879. [Google Scholar] [PubMed]
- Yang, C.; Wang, X.; Li, S.; Zhu, X.; Yu, Y.; Zhang, S. Combined analysis of transcriptomics with metabolomics provides insights into the resistance mechanism in winter jujube using L-Methionine. Plant Physiol. Biochem. 2024, 214, 108951. [Google Scholar] [CrossRef]
- Kumar, V. Evaluation of different germplasms/cultivars of tomato against early blight (Alternaria solani) in field conditions and by artificial inoculation method: A review article. Open Access Libr. J. 2015, 2, 1. [Google Scholar] [CrossRef]
- Chaerani, R.; Groenwold, R.; Stam, P.; Voorrips, R.E. Assessment of early blight (Alternaria solani) resistance in tomato using a droplet inoculation method. J. Gen. Plant Pathol. 2007, 73, 96–103. [Google Scholar] [CrossRef]
- Li, C.; Dong, N.; Shen, L.; Lu, M.; Zhai, J.; Zhao, Y.; Chen, L.; Wan, Z.; Liu, Z.; Ren, H. Genome-wide identification and expression profile of YABBY genes in Averrhoa carambola. PeerJ 2022, 10, e12558. [Google Scholar] [CrossRef]
- Kerékgyártó, M.; Kerekes, T.; Tsai, E.; Amirkhanian, V.D.; Guttman, A. Light-emitting diode induced fluorescence (LED-IF) detection design for a pen-shaped cartridge based single capillary electrophoresis system. Electrophoresis 2012, 33, 2752–2758. [Google Scholar] [CrossRef]
- Belyavsky, A.; Vinogradova, T.; Rajewsky, K. PCR-based cDNA library construction: General cDNA libraries at the level of a few cells. Nucleic Acids Res. 1989, 17, 2919–2932. [Google Scholar] [CrossRef]
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression of RNA-Seq data at the gene level–the DESeq package. Heidelb. Ger. Eur. Mol. Biol. Lab. 2012, 10, f1000research. [Google Scholar]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 2022, 39, btac757. [Google Scholar] [CrossRef]
- Yang, I.S.; Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genom. Inf. 2015, 13, 119–125. [Google Scholar] [CrossRef]
- Morgan, L.; Feller, J.; Finnegan, P. Exploring value networks: Theorising the creation and capture of value with open source software. Eur. J. Inf. Syst. 2013, 22, 569–588. [Google Scholar] [CrossRef]
- Helmig, S.; Hadzaad, B.; Döhrel, J.; Schneider, J. Relative quantification of Cytochrome P450 1B1 gene expression in peripheral leukocytes using lightcycler. Cancer Genom. Proteom. 2009, 6, 13–17. [Google Scholar]
- Clarke, K.; Gorley, R. PRIMER: Getting Started with v6; PRIMER-E Ltd.: Plymouth, UK, 2005; Volume 931, p. 932. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Li, J.; Shen, H.; Gulimila, R.; Jiang, Y.; Sun, H.; Wu, Y.; Xing, B.; Yang, R.; Liu, Y. Transcriptome Analysis of Potato (Solanum tuberosum L.) Seedlings with Varying Resistance Levels Reveals Diverse Molecular Pathways in Early Blight Resistance. Plants 2025, 14, 2422. https://doi.org/10.3390/plants14152422
Li J, Li J, Shen H, Gulimila R, Jiang Y, Sun H, Wu Y, Xing B, Yang R, Liu Y. Transcriptome Analysis of Potato (Solanum tuberosum L.) Seedlings with Varying Resistance Levels Reveals Diverse Molecular Pathways in Early Blight Resistance. Plants. 2025; 14(15):2422. https://doi.org/10.3390/plants14152422
Chicago/Turabian StyleLi, Jiangtao, Jie Li, Hongfei Shen, Rehemutula Gulimila, Yinghong Jiang, Hui Sun, Yan Wu, Binde Xing, Ruwei Yang, and Yi Liu. 2025. "Transcriptome Analysis of Potato (Solanum tuberosum L.) Seedlings with Varying Resistance Levels Reveals Diverse Molecular Pathways in Early Blight Resistance" Plants 14, no. 15: 2422. https://doi.org/10.3390/plants14152422
APA StyleLi, J., Li, J., Shen, H., Gulimila, R., Jiang, Y., Sun, H., Wu, Y., Xing, B., Yang, R., & Liu, Y. (2025). Transcriptome Analysis of Potato (Solanum tuberosum L.) Seedlings with Varying Resistance Levels Reveals Diverse Molecular Pathways in Early Blight Resistance. Plants, 14(15), 2422. https://doi.org/10.3390/plants14152422