
Integrative Population Genomics Reveals Niche Differentiation and Gene Flow in Chinese Sclerophyllous Oaks (Quercus Sect. Ilex)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification and Development of SNP Sites
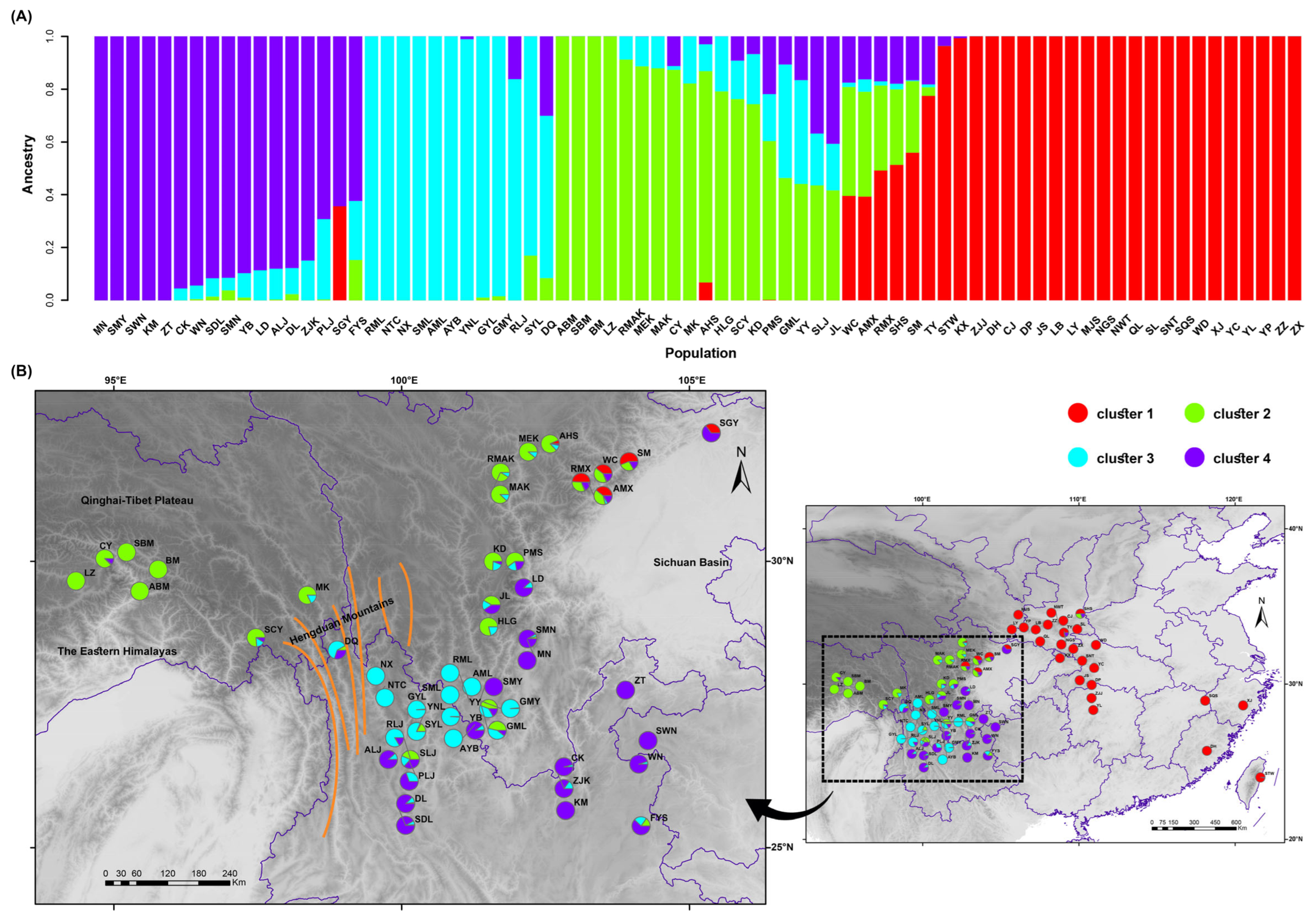
2.2. Population Genetic Structure
2.3. Phylogenetic Relationships
2.4. Gene Flow and Introgression
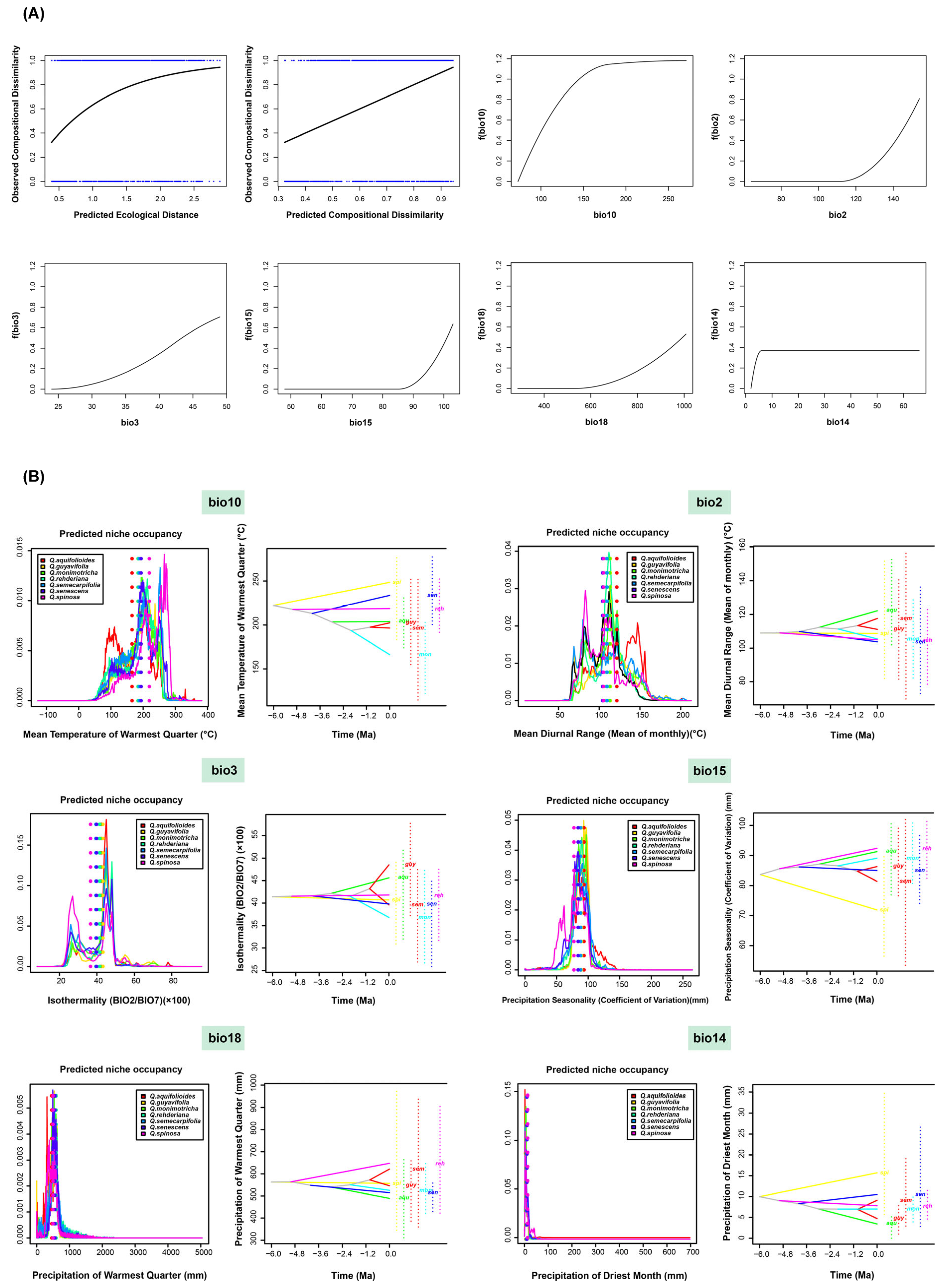
2.5. Ecological Niche Differentiation and Ancestral Reconstruction
3. Discussion
3.1. Genetic Structure and Phylogenetic Relationships
3.2. Niche Differentiation and Adaptation
4. Materials and Methods
4.1. Sample Materials
4.2. DNA Extraction and Detection
4.3. Library Preparation for SLAF Sequencing
4.4. Development of Consistent SNP Loci
4.5. Analysis of Genetic Structure
4.6. Molecular Phylogenetic Tree
4.7. Gene Flow and Introgression Detection
4.8. Ecological Niche Differentiation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodríguez-Rajo, F.J.; Méndez, J.; Jato, V. Factors Affecting Pollination Ecology of Quercus Anemophilous Species in North-West Spain. Bot. J. Linn. Soc. 2005, 149, 283–297. [Google Scholar] [CrossRef]
- Xu, X.T. Global Diversity Patterns of Quercus and Their Responses to Climate Change. Ph.D. Thesis, Peking University, Beijing, China, 2013. [Google Scholar]
- Nixon, K.C. The Genus Quercus in Mexico. Biol. Divers. Mex. 1993, 447–458. [Google Scholar]
- Kremer, A.; Delcamp, A.; Lesur, I.; Wagner, S.; Christian, R.; Guichoux, E.; Leroy, T. Whole-Genome Screening for near-Diagnostic Genetic Markers for White Oak Species Identification in Europe. bioRxiv 2023. [Google Scholar]
- Reutimann, O.; Dauphin, B.; Baltensweiler, A.; Gugerli, F.; Kremer, A.; Rellstab, C. Abiotic Factors Predict Taxonomic Composition and Genetic Admixture in Populations of Hybridizing White Oak Species (Quercus Sect. Quercus) on Regional Scale. Tree Genet. Genomes 2023, 19, 22. [Google Scholar] [CrossRef]
- Cannon, C.H.; Petit, R.J. The Oak Syngameon: More than the Sum of Its Parts. New Phytol. 2020, 226, 978–983. [Google Scholar] [CrossRef]
- Manos, P.S.; Hipp, A.L. An Updated Infrageneric Classification of the North American Oaks (Quercus Subgenus Quercus): Review of the Contribution of Phylogenomic Data to Biogeography and Species Diversity. Forests 2021, 12, 786. [Google Scholar] [CrossRef]
- Morales-Saldaña, S.; Hipp, A.L.; Valencia-Ávalos, S.; Hahn, M.; González-Elizondo, M.S.; Gernandt, D.S.; Pham, K.K.; Oyama, K.; González-Rodríguez, A. Divergence and Reticulation in the Mexican White Oaks: Ecological and Phylogenomic Evidence on Species Limits and Phylogenetic Networks in the Quercus laeta Complex (Fagaceae). Ann. Bot. 2024, 133, 1007–1024. [Google Scholar] [CrossRef]
- Kapoor, B.; Jenkins, J.; Schmutz, J.; Zhebentyayeva, T.; Kuelheim, C.; Coggeshall, M.; Heim, C.; Lasky, J.R.; Leites, L.; Islam-Faridi, N.; et al. A Haplotype-Resolved Chromosome-Scale Genome for Quercus rubra L. Provides Insights into the Genetics of Adaptive Traits for Red Oak Species. G3 Genes Genomes Genet. 2023, 13, jkad209. [Google Scholar] [CrossRef]
- Cao, M.; Zhou, Z.-K. Pollen Morphology and Its Systematic Significance of the Quercus from China. Guihaia 2002, 22, 14–18. [Google Scholar]
- Feng, Q.; Shi, Z.; Dong, L.; Liu, S. The Response of Functional Traits of Quercus Species to Meteorological Factors in Temperate Zone of NSTEC. Acta Ecol. Sin. 2010, 30, 5781–5789. [Google Scholar]
- Yang, Q.S.; Chen, W.Y.; Xia, K.; Zhou, Z.K. Climatic Envelope of Evergreen Sclerophyllous Oaks and Their Present Distribution in the Eastern Himalaya and Hengduan Mountains. J. Syst. Evol. 2009, 47, 183–190. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Z.; Rahbek, C.; Lessard, J.P.; Fang, J. Evolutionary History Influences the Effects of Water-Energy Dynamics on Oak Diversity in Asia. J. Biogeogr. 2013, 40, 2146–2155. [Google Scholar] [CrossRef]
- Yuan, S.; Shi, Y.; Zhou, B.F.; Liang, Y.Y.; Chen, X.Y.; An, Q.Q.; Fan, Y.R.; Shen, Z.; Ingvarsson, P.K.; Wang, B. Genomic Vulnerability to Climate Change in Quercus acutissima, a Dominant Tree Species in East Asian Deciduous Forests. Mol. Ecol. 2023, 32, 1639–1655. [Google Scholar] [CrossRef]
- Ai, W.; Liu, Y.; Mei, M.; Zhang, X.; Tan, E.; Liu, H.; Han, X.; Zhan, H.; Lu, X. A Chromosome-Scale Genome Assembly of the Mongolian Oak (Quercus mongolica). Mol. Ecol. Resour. 2022, 22, 2396–2410. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.Y.; Shi, Y.; Yuan, S.; Zhou, B.F.; Chen, X.Y.; An, Q.Q.; Ingvarsson, P.K.; Plomion, C.; Wang, B. Linked Selection Shapes the Landscape of Genomic Variation in Three Oak Species. New Phytol. 2022, 233, 555–568. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, B.F.; Liang, Y.Y.; Wang, B. Linked Selection and Recombination Rate Generate both Shared and Lineage-Specific Genomic Islands of Divergence in Two Independent Quercus Species Pairs. J. Syst. Evol. 2023, 62, 505–519. [Google Scholar] [CrossRef]
- Yang, J.; Di, X.; Meng, X.; Feng, L.; Liu, Z.; Zhao, G. Phylogeography and Evolution of Two Closely Related Oak Species (Quercus) from North and Northeast China. Tree Genet. Genomes 2016, 12, 89. [Google Scholar] [CrossRef]
- Shi, X.; Wen, Q.; Cao, M.; Guo, X.; Xu, L.A. Genetic Diversity and Structure of Natural Quercus variabilis Population in China as Revealed by Microsatellites Markers. Forests 2017, 8, 495. [Google Scholar] [CrossRef]
- Jiang, X.L.; Hipp, A.L.; Deng, M.; Su, T.; Zhou, Z.K.; Yan, M.X. East Asian Origins of European Holly Oaks (Quercus Section Ilex Loudon) via the Tibet-Himalaya. J. Biogeogr. 2019, 46, 2203–2214. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, C.; Wang, Q.; Jiang, Y.; Qin, L. Germplasm Resources of Oaks (Quercus L.) in China: Utilization and Prospects. Biology 2023, 12, 76. [Google Scholar] [CrossRef]
- Hipp, A.L.; Manos, P.S.; Hahn, M.; Avishai, M.; Bodénès, C.; Cavender-Bares, J.; Crowl, A.A.; Deng, M.; Denk, T.; Fitz-Gibbon, S.; et al. Genomic Landscape of the Global Oak Phylogeny. New Phytol. 2020, 226, 1198–1212. [Google Scholar] [CrossRef]
- Du, F.K.; Hou, M.; Wang, W.; Mao, K.; Hampe, A. Phylogeography of Quercus aquifolioides Provides Novel Insights into the Neogene History of a Major Global Hotspot of Plant Diversity in South-West China. J. Biogeogr. 2017, 44, 294–307. [Google Scholar] [CrossRef]
- Meng, H.H.; Su, T.; Gao, X.Y.; Li, J.; Jiang, X.L.; Sun, H.; Zhou, Z.K. Warm–Cold Colonization: Response of Oaks to Uplift of the Himalaya–Hengduan Mountains. Mol. Ecol. 2017, 26, 3276–3294. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.M.; Yang, J.; Yue, M.; Zhao, G.F. Speciation Patterns of Related Species under the Hybrid Zone: A Case Study of Three Sclerophyllous Oaks in the East Himalaya–Hengduan Mountains. Mol. Ecol. 2023, 32, 4610–4626. [Google Scholar] [CrossRef]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-Seq: An Efficient Method of Large-Scale De Novo SNP Discovery and Genotyping Using High-Throughput Sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Ju, M.M.; Feng, L.; Yang, J.; Yang, Y.C.; Chen, X.D.; Zhao, G.F. Evaluating Population Genetic Structure and Demographic History of Quercus Spinosa (Fagaceae) Based on Specific Length Amplified Fragment Sequencing. Front. Genet. 2019, 10, 965. [Google Scholar] [CrossRef] [PubMed]
- Schoener, T.W. The Anolis Lizards of Bimini: Resource Partitioning in a Complex Fauna. Ecology 1968, 49, 704–726. [Google Scholar] [CrossRef]
- Rödder, D.; Engler, J.O. Quantitative Metrics of Overlaps in Grinnellian Niches: Advances and Possible Drawbacks. Glob. Ecol. Biogeogr. 2011, 20, 915–927. [Google Scholar] [CrossRef]
- Wu, Z.Y.; Wu, S.G. A Proposal for a New Floristic Kingdom (Realm): The E. Asiatic Kingdom, Its Delimitation and Characteristics. In Floristic Characteristics and Diversity of East Asian Plants; Zhang, A.L., Wu, S.G., Eds.; China Higher Education & Springer Asia: Beijing, China, 1996; pp. 3–42. [Google Scholar]
- Zhang, D.; Li, F.; Bian, J. Eco-Environmental Effects of the Qinghai-Tibet Plateau Uplift during the Quaternary in China. Environ. Geol. 2000, 39, 1352–1358. [Google Scholar] [CrossRef]
- Ye, J.W.; Zhang, Y.; Wang, X.J. Phylogeographic Breaks and the Mechanisms of Their Formation in the Sino-Japanese Floristic Region. Chin. J. Plant Ecol. 2017, 41, 1003–1019. [Google Scholar]
- Yan, H.F.; Zhang, C.Y.; Wang, F.Y.; Hu, C.M.; Ge, X.J.; Hao, G. Population Expanding with the Phalanx Model and Lineages Split by Environmental Heterogeneity: A Case Study of Primula obconica in Subtropical China. PLoS ONE 2012, 7, e41315. [Google Scholar] [CrossRef]
- Cao, Y.N.; Comes, H.P.; Sakaguchi, S.; Chen, L.Y.; Qiu, Y.X. Evolution of East Asia’s Arcto-Tertiary Relict Euptelea (Eupteleaceae) Shaped by Late Neogene Vicariance and Quaternary Climate Change. BMC Evol. Biol. 2016, 16, 66. [Google Scholar] [CrossRef]
- Ye, J.W.; Zhang, Y.; Wang, X.J. Phylogeographic History of Broad-Leaved Forest Plants in Subtropical China. Acta Ecol. Sin. 2017, 37, 5894–5904. [Google Scholar] [CrossRef]
- Xu, J.; Deng, M.; Jiang, X.L.; Westwood, M.; Song, Y.G.; Turkington, R. Phylogeography of Quercus glauca (Fagaceae), a Dominant Tree of East Asian Subtropical Evergreen Forests, Based on Three Chloroplast DNA Interspace Sequences. Tree Genet. Genomes 2015, 11, 805. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, H.; Huang, H.; Vargas-Mendoza, C.F. Chloroplast Diversity and Population Differentiation of Castanopsis fargesii (Fagaceae): A Dominant Tree Species in Evergreen Broad-Leaved Forest of Subtropical China. Tree Genet. Genomes 2014, 10, 1531–1539. [Google Scholar] [CrossRef]
- Shi, M.M.; Michalski, S.G.; Welk, E.; Chen, X.Y.; Durka, W. Phylogeography of a Widespread Asian Subtropical Tree: Genetic East-West Differentiation and Climate Envelope Modelling Suggest Multiple Glacial Refugia. J. Biogeogr. 2014, 41, 1710–1720. [Google Scholar] [CrossRef]
- Li, J.; Fang, X. Uplift of the Tibetan Plateau and Environmental Changes. Chin. Sci. Bull. 1999, 44, 2117–2124. [Google Scholar] [CrossRef]
- Mulch, A.; Chamberlain, C.P. The Rise and Growth of Tibet. Nature 2006, 439, 670–671. [Google Scholar] [CrossRef]
- Favre, A.; Päckert, M.; Pauls, S.U.; Jähnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The Role of the Uplift of the Qinghai-Tibetan Plateau for the Evolution of Tibetan Biotas. Biol. Rev. Camb. Philos. Soc. 2015, 90, 236–253. [Google Scholar] [CrossRef]
- He, K.; Jiang, X. Sky Islands of Southwest China I: An Overview of Phylogeographic Patterns. Chin. Sci. Bull. 2014, 59, 585–597. [Google Scholar] [CrossRef]
- Stokstad, E. Mountains and Monsoons Created Tibetan Biodiversity. Science 2020, 369, 493. [Google Scholar] [CrossRef]
- Liao, Z.; Nobis, M.P.; Xiong, Q.; Tian, X.; Wu, X.; Pan, K.; Zhang, A.; Wang, Y.; Zhang, L. Potential Distributions of Seven Sympatric Sclerophyllous Oak Species in Southwest China Depend on Climatic, Non-Climatic, and Independent Spatial Drivers. Ann. For. Sci. 2021, 78, 5. [Google Scholar] [CrossRef]
- Teeter, K.C.; Thibodeau, L.M.; Gompert, Z.; Buerkle, C.A.; Nachman, M.W.; Tucker, P.K. The Variable Genomic Architecture of Isolation Between Hybridizing Species of House Mice. Evol. Int. J. Org. Evol. 2010, 64, 472–485. [Google Scholar] [CrossRef]
- Teeter, K.C.; Payseur, B.A.; Harris, L.W.; Bakewell, M.A.; Thibodeau, L.M.; O Brien, J.E.; Krenz, J.G.; Sans-Fuentes, M.A.; Nachman, M.W.; Tucker, P.K. Genome-Wide Patterns of Gene Flow across a House Mouse Hybrid Zone. Genome Res. 2008, 18, 67–76. [Google Scholar] [CrossRef]
- Ryan, S.F.; Fontaine, M.C.; Scriber, J.M.; Pfrender, M.E.; O’Neil, S.T.; Hellmann, J.J. Patterns of Divergence across the Geographic and Genomic Landscape of a Butterfly Hybrid Zone Associated with a Climatic Gradient. Mol. Ecol. 2017, 26, 4725–4742. [Google Scholar] [CrossRef]
- Sun, Y.; Abbott, R.J.; Lu, Z.; Mao, K.; Zhang, L.; Wang, X.; Ru, D.; Liu, J. Reticulate Evolution within a Spruce (Picea) Species Complex Revealed by Population Genomic Analysis. Evolution 2018, 72, 2669–2681. [Google Scholar] [CrossRef]
- Yacine, A.; Bouras, F. Self-and Cross-Pollination Effects on Pollen Tube Growth and Seed Set in Holm Oak Quercus ilex L (Fagaceae). Ann. Des Sci. For. 1997, 54, 447–462. [Google Scholar] [CrossRef]
- Ortego, J.; Gugger, P.F.; Riordan, E.C.; Sork, V.L. Influence of Climatic Niche Suitability and Geographical Overlap on Hybridization Patterns among Southern Californian Oaks. J. Biogeogr. 2014, 41, 1895–1908. [Google Scholar] [CrossRef]
- Ortego, J.; Gugger, P.F.; Sork, V.L. Climatically Stable Landscapes Predict Patterns of Genetic Structure and Admixture in the Californian Canyon Live Oak. J. Biogeogr. 2015, 42, 328–338. [Google Scholar] [CrossRef]
- Ortego, J.; Gugger, P.F.; Sork, V.L. Impacts of Human-Induced Environmental Disturbances on Hybridization Between Two Ecologically Differentiated Californian Oak Species. New Phytol. 2017, 213, 942–955. [Google Scholar] [CrossRef]
- Ortego, J.; Noguerales, V.; Gugger, P.F.; Sork, V.L. Evolutionary and Demographic History of the Californian Scrub White Oak Species Complex: An Integrative Approach. Mol. Ecol. 2015, 24, 6188–6208. [Google Scholar] [CrossRef]
- Guichoux, E.; Garnier-Gere, P.; Lagache, L.; Lang, T.; Boury, C.; Petit, R.J. Outlier Loci Highlight the Direction of Introgression in Oaks. Mol. Ecol. 2013, 22, 450–462. [Google Scholar] [CrossRef]
- Hegarty, M.J.; Hiscock, S.J. Hybrid Speciation in Plants: New Insights from Molecular Studies. New Phytol. 2005, 165, 411–423. [Google Scholar] [CrossRef]
- Mallet, J. Hybrid Speciation. Nature 2007, 446, 279–283. [Google Scholar] [CrossRef]
- Janke, A. Divergence with Genetic Exchange.—M.L. Arnold. Syst. Biol. 2016, 65, 941–942. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Hu, Q.; Li, J.; Sun, Y.; Zhang, L.; Abbott, R.J.; Liu, J.; Mao, K. Ancient Introgression Drives Adaptation to Cooler and Drier Mountain Habitats in a Cypress Species Complex. Commun. Biol. 2019, 2, 213. [Google Scholar] [CrossRef]
- Pluess, A.R.; Sork, V.L.; Dolan, B.; Davis, F.W.; Grivet, D.; Merg, K.; Papp, J.; Smouse, P.E. Short Distance Pollen Movement in a Wind-Pollinated Tree, Quercus lobata (Fagaceae). For. Ecol. Manag. 2009, 258, 735–744. [Google Scholar] [CrossRef]
- Fu, R.; Zhu, Y.; Liu, Y.; Feng, Y.; Lu, R.S.; Li, Y.; Li, P.; Kremer, A.; Lascoux, M.; Chen, J. Genome-Wide Analyses of Introgression Between Two Sympatric Asian Oak Species. Nat. Ecol. Evol. 2022, 6, 924–935. [Google Scholar] [CrossRef]
- Van Valen, L. Ecological Species, Multispecies, and Oaks. Taxon 1976, 25, 233–239. [Google Scholar] [CrossRef]
- Maire, V.; Gross, N.; Börger, L.; Proulx, R.; Wirth, C.; Pontes, L.D.S.; Soussana, J.F.; Louault, F. Habitat Filtering and Niche Differentiation Jointly Explain Species Relative Abundance Within Grassland Communities Along Fertility and Disturbance Gradients. New Phytol. 2012, 196, 497–509. [Google Scholar] [CrossRef]
- Kim, D.; Ohr, S. Coexistence of Plant Species Under Harsh Environmental Conditions: An Evaluation of Niche Differentiation and Stochasticity Along Salt Marsh Creeks. J. Ecol. Environ. 2020, 44, 19. [Google Scholar] [CrossRef]
- Obayomi, O.; Seyoum, M.M.; Ghazaryan, L.; Tebbe, C.C.; Murase, J.; Bernstein, N.; Gillor, O. Soil Texture and Properties Rather than Irrigation Water Type Shape the Diversity and Composition of Soil Microbial Communities. Appl. Soil Ecol. 2021, 161, 103834. [Google Scholar] [CrossRef]
- Chai, Z.; Fan, D.; Wang, D. Environmental Factors and Underlying Mechanisms of Tree Community Assemblages of Pine-Oak Mixed Forests in the Qinling Mountains, China. J. Plant Biol. 2016, 59, 347–357. [Google Scholar] [CrossRef]
- He, Y.; Liang, S.; Jiang, Y.; Ning, W. The Relative Importance of Niche and Neutral Processes for the Community Assembly of Subtropical Karst Forest Communities at Different Spatial Scales. Forests 2022, 13, 1930. [Google Scholar] [CrossRef]
- Michael, H.; Freeman, J. Organizational Ecology; Harvard University Press: Cambridge, MA, USA, 1989. [Google Scholar]
- Chen, D.; Liao, J.; Bearup, D.; Li, Z. Habitat Heterogeneity Mediates Effects of Individual Variation on Spatial Species Coexistence. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192436. [Google Scholar] [CrossRef]
- Kyogoku, D.; Kokko, H. Species Coexist More Easily If Reinforcement Is Based on Habitat Preferences than on Species Recognition. J. Anim. Ecol. 2020, 89, 2605–2616. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimum Sample Sizes for Population Genomics: An Empirical Study from an Amazonian Plant Species. Mol. Ecol. Resour. 2017, 17, 1136–1147. [Google Scholar] [CrossRef]
- Chen, L.Y.; Song, M.S.; Zha, H.G.; Li, Z.M. A Modified Protocol for Plant Genome Dna Extraction. Plant Divers. 2014, 36, 375–380. [Google Scholar]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the Miseq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Chifman, J.; Kubatko, L. Quartet Inference from SNP Data under the Coalescent Model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP * Phylogenetic Analysis Using Parsimony * (and Other Methods), Version 4.0; Sinauer Associates: Sunderland, MA, USA, 2002; Volume 2002. [Google Scholar]
- Leache, A.D.; Chavez, A.S.; Jones, L.N.; Grummer, J.A.; Gottscho, A.D.; Linkem, C.W. Phylogenomics of Phrynosomatid Lizards: Conflicting Signals from Sequence Capture versus Restriction Site Associated DNA Sequencing. Genome Biol. Evol. 2015, 7, 706–719. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast D-Statistics and Related Admixture Evidence from VCF Files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum Entropy Modeling of Species Geographic Distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- GBIF.org (2023), GBIF Home Page. Available online: https://www.gbif.org (accessed on 1 October 2023).
- Warren, D.L.; Glor, R.E.; Turelli, M. Environmental Niche Equivalency versus Conservatism: Quantitative Approaches to Niche Evolution. Evol. Int. J. Org. Evol. 2008, 62, 2868–2883. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.E.K.; Smith, S.A.; Flynn, R.S.; Donoghue, M.J. Climate, Niche Evolution, and Diversification of The “bird-Cage” evening Primroses (Oenothera, Sections Anogra and Kleinia). Am. Nat. 2009, 173, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, S.; Manion, G.; Elith, J.; Richardson, K. Using Generalized Dissimilarity Modelling to Analyse and Predict Patterns of Beta Diversity in Regional Biodiversity Assessment. Divers. Distrib. 2007, 13, 252–264. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, M.-M.; Yue, M.; Zhao, G.-F. Integrative Population Genomics Reveals Niche Differentiation and Gene Flow in Chinese Sclerophyllous Oaks (Quercus Sect. Ilex). Plants 2025, 14, 2403. https://doi.org/10.3390/plants14152403
Ju M-M, Yue M, Zhao G-F. Integrative Population Genomics Reveals Niche Differentiation and Gene Flow in Chinese Sclerophyllous Oaks (Quercus Sect. Ilex). Plants. 2025; 14(15):2403. https://doi.org/10.3390/plants14152403
Chicago/Turabian StyleJu, Miao-Miao, Ming Yue, and Gui-Fang Zhao. 2025. "Integrative Population Genomics Reveals Niche Differentiation and Gene Flow in Chinese Sclerophyllous Oaks (Quercus Sect. Ilex)" Plants 14, no. 15: 2403. https://doi.org/10.3390/plants14152403
APA StyleJu, M.-M., Yue, M., & Zhao, G.-F. (2025). Integrative Population Genomics Reveals Niche Differentiation and Gene Flow in Chinese Sclerophyllous Oaks (Quercus Sect. Ilex). Plants, 14(15), 2403. https://doi.org/10.3390/plants14152403