
Construction of Ancestral Chromosomes in Gymnosperms and the Application in Comparative Genomic Analysis

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
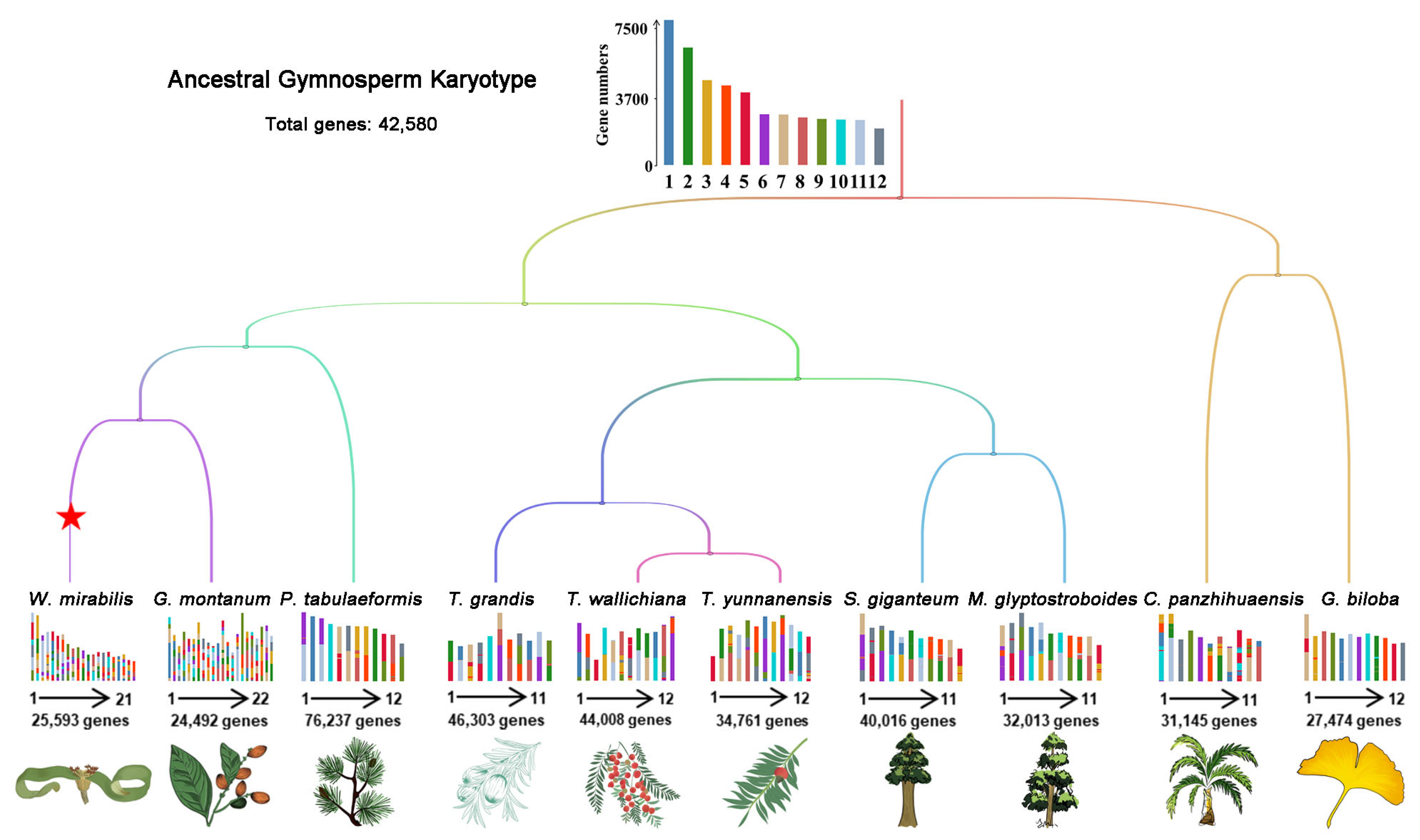
2.1. Reconstruction of Ancestral Gymnosperm Karyotype
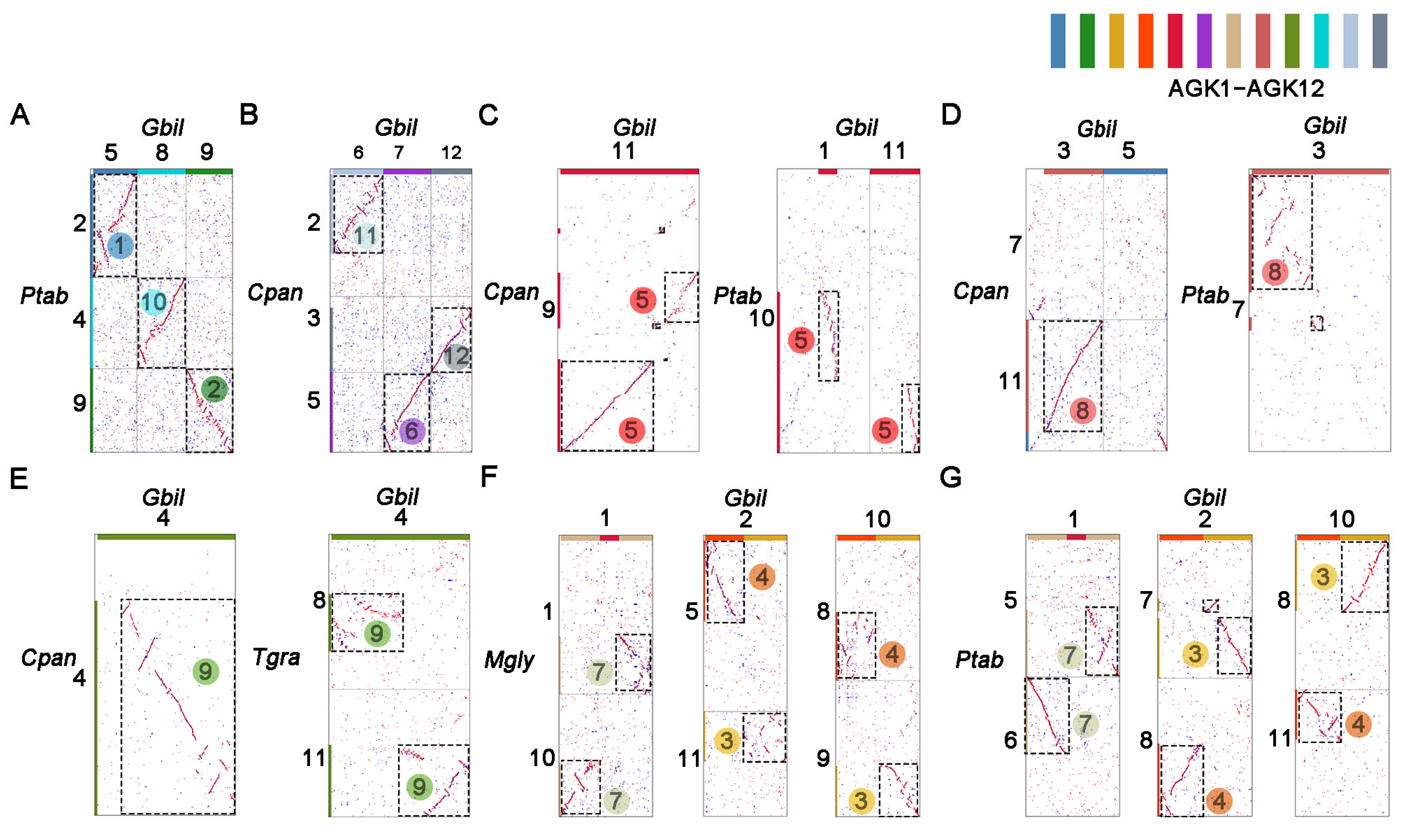
2.2. Karyotype Mapping and Collinearity Analyses of Gymnosperms
2.3. Distribution of Duplicated Blocks in Species with Multiplied Chromosome Numbers
2.4. Enrichment of LINE1 Elements in Retained Genomic Blocks in the W. mirabilis Genome
2.5. Characteristics of Blocks in Angiosperms That Are Collinear with AGK
3. Discussion
3.1. Ancestral Chromosomes of Gymnosperms
3.2. Chromosome Stability and Chromosome Numbers
3.3. Potential Roles of Repetitive Sequences in Maintaining Chromosome Stability
4. Materials and Methods
4.1. Data Sources
4.2. Reconstruction of AGK and Karyotype Projections
4.3. Collinearity Analyses
4.4. Repeat Annotation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, K.; Wang, X.; Cheng, F. Plant Polyploidy: Origin, Evolution, and Its Influence on Crop Domestication. Hortic. Plant J. 2019, 5, 231–239. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, S.Z.; Liu, Y.L.; Zhao, B.Y.; Yu, D.; Zhong, M.C.; Jiang, X.D.; Cui, W.H.; Zhao, J.X.; Qiu, J.; et al. Genomes of Meniocus linifolius and Tetracme quadricornis reveal the ancestral karyotype and features of core Brassicaceae. Plant Commun. 2024, 5, 100878. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.B.; Chen, R.; Liao, K.X.; Li, H.; Xu, G.B.; Jiang, X.L. Karyotype and LTR-RTs analysis provide insights into oak genomic evolution. BMC Genom. 2024, 25, 328. [Google Scholar] [CrossRef] [PubMed]
- Tkach, N.; Winterfeld, G.; Roeser, M. Genome sizes of grasses (Poaceae), chromosomal evolution, paleogenomics and the ancestral grass karyotype (AGK). Plant Syst. Evol. 2025, 311, 4. [Google Scholar] [CrossRef]
- Emonet, A.; Awad, M.; Tikhomirov, N.; Vasilarou, M.; Perez-Anton, M.; Gan, X.; Novikova, P.Y.; Hay, A. Polyploid genome assembly of Cardamine chenopodiifolia. GigaByte 2024, 2024, gigabyte145. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Zhang, Q.; Huang, J.; Zhang, S.; Yao, W.; Yu, Z.; Deng, Z.; Yu, J.; Kong, W.; Yu, X.; et al. A chromosomal-scale genome assembly of modern cultivated hybrid sugarcane provides insights into origination and evolution. Nat. Commun. 2024, 15, 3041. [Google Scholar] [CrossRef]
- Murat, F.; Xu, J.H.; Tannier, E.; Abrouk, M.; Guilhot, N.; Pont, C.; Messing, J.; Salse, J. Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution. Genome Res. 2010, 20, 1545–1557. [Google Scholar] [CrossRef]
- Ma, P.F.; Liu, Y.L.; Guo, C.; Jin, G.; Guo, Z.H.; Mao, L.; Yang, Y.Z.; Niu, L.Z.; Wang, Y.J.; Clark, L.G.; et al. Genome assemblies of 11 bamboo species highlight diversification induced by dynamic subgenome dominance. Nat. Genet. 2024, 56, 710–720. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Sun, P.; Zhu, M.; Wang, D.; Lu, Z.; Hu, H.; Xu, R.; Zhang, J.; Ma, J.; et al. A high-quality Buxus austro-yunnanensis (Buxales) genome provides new insights into karyotype evolution in early eudicots. BMC Biol. 2022, 20, 216. [Google Scholar] [CrossRef]
- Shao, L.; Jin, S.; Chen, J.; Yang, G.; Fan, R.; Zhang, Z.; Deng, Q.; Han, J.; Ma, X.; Dong, Z.; et al. High-quality genomes of Bombax ceiba and Ceiba pentandra provide insights into the evolution of Malvaceae species and differences in their natural fiber development. Plant Commun. 2024, 5, 100832. [Google Scholar] [CrossRef]
- Murat, F.; Armero, A.; Pont, C.; Klopp, C.; Salse, J. Reconstructing the genome of the most recent common ancestor of flowering plants. Nat. Genet. 2017, 49, 490–496. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, Y.; Wang, Z.; Bao, S.; Feng, Y.; Wang, J.; Yu, Z.; Long, F.; Xiao, Z.; Hao, Y.; et al. Two-step model of paleohexaploidy, ancestral genome reshuffling and plasticity of heat shock response in Asteraceae. Hortic. Res. 2023, 10, uhad073. [Google Scholar] [CrossRef]
- Carvalho, M.R.; Jaramillo, C.; de la Parra, F.; Caballero-Rodriguez, D.; Herrera, F.; Wing, S.; Turner, B.L.; D’Apolito, C.; Romero-Baez, M.; Narvaez, P.; et al. Extinction at the end-Cretaceous and the origin of modern Neotropical rainforests. Science 2021, 372, 63–68. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Fan, G.; Yin, P.P.; Sun, S.; Li, N.; Hong, X.; Hu, G.; Zhang, H.; Zhang, F.M.; Han, J.D.; et al. Resequencing 545 ginkgo genomes across the world reveals the evolutionary history of the living fossil. Nat. Commun. 2019, 10, 4201. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ferguson, D.K.; Liu, B.; Mao, K.S.; Gao, L.M.; Zhang, S.Z.; Wan, T.; Rushforth, K.; Zhang, Z.X. Recent advances on phylogenomics of gymnosperms and a new classification. Plant Divers. 2022, 44, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Stull, G.W.; Qu, X.J.; Parins-Fukuchi, C.; Yang, Y.Y.; Yang, J.B.; Yang, Z.Y.; Hu, Y.; Ma, H.; Soltis, P.S.; Soltis, D.E.; et al. Gene duplications and phylogenomic conflict underlie major pulses of phenotypic evolution in gymnosperms. Nat. Plants 2021, 7, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Sauquet, H.; von Balthazar, M.; Magallon, S.; Doyle, J.A.; Endress, P.K.; Bailes, E.J.; Barroso de Morais, E.; Bull-Herenu, K.; Carrive, L.; Chartier, M.; et al. The ancestral flower of angiosperms and its early diversification. Nat. Commun. 2017, 8, 16047. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Ran, J.H.; Shen, T.T.; Wang, M.M.; Wang, X.Q. Phylogenomics resolves the deep phylogeny of seed plants and indicates partial convergent or homoplastic evolution between Gnetales and angiosperms. Proc. Biol. Sci./R. Soc. 2018, 285, 20181012. [Google Scholar] [CrossRef]
- Pandey, B.; Pan, K.; Dakhil, M.A.; Liao, Z.; Timilsina, A.; Khanal, M.; Zhang, L. Contrasting Gymnosperm Diversity Across an Elevation Gradient in the Ecoregion of China: The Role of Temperature and Productivity. Front. Ecol. Evol. 2021, 9, 679439. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Li, L.; Yang, T.; Dong, S.; Wei, T.; Wu, S.; Liu, Y.; Gong, Y.; Feng, X.; et al. The Cycas genome and the early evolution of seed plants. Nat. Plants 2022, 8, 389–401. [Google Scholar] [CrossRef]
- Sanderson, B.J.; Feng, G.; Hu, N.; Carlson, C.H.; Smart, L.B.; Keefover-Ring, K.; Yin, T.; Ma, T.; Liu, J.; DiFazio, S.P.; et al. Sex determination through X-Y heterogamety in Salix nigra. Heredity 2021, 126, 630–639. [Google Scholar] [CrossRef]
- Song, C.; Fu, F.; Yang, L.; Niu, Y.; Tian, Z.; He, X.; Yang, X.; Chen, J.; Sun, W.; Wan, T.; et al. Taxus yunnanensis genome offers insights into gymnosperm phylogeny and taxol production. Commun. Bology 2021, 4, 1203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wang, X.; Liu, X.; Zhu, X.; Li, Z.; Chu, H.; Wang, Q.; Lou, Q.; Cai, B.; Yang, Y.; et al. Chromosome-level genome of Himalayan yew provides insights into the origin and evolution of the paclitaxel biosynthetic pathway. Mol. Plant 2021, 14, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Gou, J.; Liao, Q.; Li, Y.; Zhou, Q.; Bi, G.; Li, C.; Du, R.; Wang, X.; Sun, T.; et al. The Taxus genome provides insights into paclitaxel biosynthesis. Nat. Plants 2021, 7, 1026–1036. [Google Scholar] [CrossRef]
- Lou, H.; Song, L.; Li, X.; Zi, H.; Chen, W.; Gao, Y.; Zheng, S.; Fei, Z.; Sun, X.; Wu, J. The Torreya grandis genome illuminates the origin and evolution of gymnosperm-specific sciadonic acid biosynthesis. Nat. Commun. 2023, 14, 1315. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Song, C.; Wen, C.; Yang, L.; Guo, Y.; Yang, X.; Shu, Z.; Li, X.; Feng, Y.; Liu, B.; et al. The Metasequoia genome and evolutionary relationships among redwoods. Plant Commun 2023, 4, 100643. [Google Scholar] [CrossRef]
- Neale, D.B.; Zimin, A.V.; Zaman, S.; Scott, A.D.; Shrestha, B.; Workman, R.E.; Puiu, D.; Allen, B.J.; Moore, Z.J.; Sekhwal, M.K.; et al. Assembled and annotated 26.5 Gbp coast redwood genome: A resource for estimating evolutionary adaptive potential and investigating hexaploid origin. G3 Genes/Genomes/Genet. 2022, 12, jkab380. [Google Scholar] [CrossRef]
- Niu, S.; Li, J.; Bo, W.; Yang, W.; Zuccolo, A.; Giacomello, S.; Chen, X.; Han, F.; Yang, J.; Song, Y.; et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 2022, 185, 204–217.e214. [Google Scholar] [CrossRef]
- Wan, T.; Liu, Z.; Leitch, I.J.; Xin, H.; Maggs-Kolling, G.; Gong, Y.; Li, Z.; Marais, E.; Liao, Y.; Dai, C.; et al. The Welwitschia genome reveals a unique biology underpinning extreme longevity in deserts. Nat. Commun. 2021, 12, 4247. [Google Scholar] [CrossRef]
- Zhang, R.G.; Liu, H.; Shang, H.Y.; Shu, H.; Liu, D.T.; Yang, H.; Jia, K.H.; Wang, X.Q.; Sun, W.B.; Zhao, W.; et al. Convergent Patterns of Karyotype Evolution Underlying Karyotype Uniformity in Conifers. Adv. Sci. 2025, 12, e2411098. [Google Scholar] [CrossRef]
- Salas-Leiva, D.E.; Meerow, A.W.; Calonje, M.; Griffith, M.P.; Francisco-Ortega, J.; Nakamura, K.; Stevenson, D.W.; Lewis, C.E.; Namoff, S. Phylogeny of the cycads based on multiple single-copy nuclear genes: Congruence of concatenated parsimony, likelihood and species tree inference methods. Ann. Bot. 2013, 112, 1263–1278. [Google Scholar] [CrossRef]
- Condamine, F.L.; Nagalingum, N.S.; Marshall, C.R.; Morlon, H. Origin and diversification of living cycads: A cautionary tale on the impact of the branching process prior in Bayesian molecular dating. BMC Evol. Biol. 2015, 15, 65. [Google Scholar] [CrossRef]
- Crisp, M.D.; Cook, L.G. Cenozoic extinctions account for the low diversity of extant gymnosperms compared with angiosperms. New Phytol. 2011, 192, 997–1009. [Google Scholar] [CrossRef]
- Nagalingum, N.S.; Marshall, C.R.; Quental, T.B.; Rai, H.S.; Little, D.P.; Mathews, S. Recent synchronous radiation of a living fossil. Science 2011, 334, 796–799. [Google Scholar] [CrossRef]
- Jiang, X.; Hu, Q.; Mei, D.; Li, X.; Xiang, L.; Al-Shehbaz, I.A.; Song, X.; Liu, J.; Lysak, M.A.; Sun, P. Chromosome fusions shaped karyotype evolution and evolutionary relationships in the model family Brassicaceae. Nat. Commun. 2025, 16, 4631. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, F.; Zhang, X.; Li, Z.; Zhao, Y.; Lohaus, R.; Chang, X.; Dong, W.; Ho, S.Y.W.; Liu, X.; et al. The water lily genome and the early evolution of flowering plants. Nature 2020, 577, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Ove, N.; Teitur Ahlgren, K.; Nicolas, D.; Mimmi, E. 1000 conifer genomes: Genome innovation, organisation and diversity. Res. Sq. 2025. [Google Scholar] [CrossRef]
- Feng, X.; Chen, Q.; Wu, W.; Wang, J.; Li, G.; Xu, S.; Shao, S.; Liu, M.; Zhong, C.; Wu, C.I.; et al. Genomic evidence for rediploidization and adaptive evolution following the whole-genome triplication. Nat. Commun. 2024, 15, 1635. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 2015, 102, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.G.; Hoerandl, E.; Zhang, Z.X.; Charlesworth, D.; He, L. Evolution of Sex-linked Genes and the Role of Pericentromeric Regions in Sex Chromosomes: Insights from Diploid Willows. Mol. Biol. Evol. 2024, 41, msae235. [Google Scholar] [CrossRef]
- Li, Z.; Baniaga, A.E.; Sessa, E.B.; Scascitelli, M.; Graham, S.W.; Rieseberg, L.H.; Barker, M.S. Early genome duplications in conifers and other seed plants. Sci. Adv. 2015, 1, e1501084. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Liu, Z.M.; Li, L.F.; Leitch, A.R.; Leitch, I.J.; Lohaus, R.; Liu, Z.J.; Xin, H.P.; Gong, Y.B.; Liu, Y.; et al. A genome for gnetophytes and early evolution of seed plants. Nat. Plants 2018, 4, 82–89. [Google Scholar] [CrossRef]
- Marchant, D.B.; Chen, G.; Cai, S.; Chen, F.; Schafran, P.; Jenkins, J.; Shu, S.; Plott, C.; Webber, J.; Lovell, J.T.; et al. Dynamic genome evolution in a model fern. Nat. Plants 2021, 8, 1038–1051. [Google Scholar] [CrossRef]
- Scott, A.D.; Stenz, N.W.; Ingvarsson, P.K.; Baum, D.A. Whole genome duplication in coast redwood (Sequoia sempervirens) and its implications for explaining the rarity of polyploidy in conifers. New Phytol. 2016, 211, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Leitch, I.J.; Bennett, M.D. Genome downsizing in polyploid plants. Biol. J. Linn. Soc. 2004, 82, 651–663. [Google Scholar] [CrossRef]
- Yingjin, T.; Taicong, T.; Shuxian, Z.; Bo, L.; Beiyi, C.; Xu, Z.; Ying, W.; Yang, X.; Binyuan, Z.; Qilai, H.; et al. Temperature regulates negative supercoils to modulate meiotic crossovers and chromosome organization. Sci. China Life Sci. 2024, 67, 2426–2443. [Google Scholar] [CrossRef]
- Farhat, P.; Hidalgo, O.; Robert, T.; Siljak-Yakovlev, S.; Leitch, I.J.; Adams, R.P.; Bou Dagher-Kharrat, M. Polyploidy in the Conifer Genus Juniperus: An Unexpectedly High Rate. Front. Plant Sci. 2019, 10, 676. [Google Scholar] [CrossRef]
- Ickert-Bond, S.M.; Sousa, A.; Min, Y.; Loera, I.; Metzgar, J.; Pellicer, J.; Hidalgo, O.; Leitch, I.J. Polyploidy in gymnosperms—Insights into the genomic and evolutionary consequences of polyploidy in Ephedra. Mol. Phylogenetics Evol. 2020, 147, 106786. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.T.; Sreedasyam, A.; Schranz, M.E.; Wilson, M.; Carlson, J.W.; Harkess, A.; Emms, D.; Goodstein, D.M.; Schmutz, J. GENESPACE tracks regions of interest and gene copy number variation across multiple genomes. Elife 2022, 11, e78526. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Steenwyk, J.L.; LaBella, A.L.; Harrison, M.C.; Groenewald, M.; Zhou, X.; Shen, X.X.; Zhao, T.; Hittinger, C.T.; et al. Contrasting modes of macro and microsynteny evolution in a eukaryotic subphylum. Curr. Biol. 2022, 32, 5335–5343. [Google Scholar] [CrossRef]
- Almeida-Silva, F.; Zhao, T.; Ullrich, K.K.; Schranz, M.E.; Van de Peer, Y. syntenet: An R/Bioconductor package for the inference and analysis of synteny networks. Bioinformatics 2023, 39, btac806. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Liu, W.; Li, J.; Xiong, S.; Jia, J.; Lin, Q.; Liu, H.; Cui, P. Evolution of Plant Genome Size and Composition. Genom. Proteom. Bioinform. 2024, 22, qzae078. [Google Scholar] [CrossRef] [PubMed]
- Scelfo, A.; Fachinetti, D. Centromere: A Trojan horse for genome stability. DNA Repair 2023, 130, 103569. [Google Scholar] [CrossRef]
- Naish, M.; Henderson, I.R. The structure, function, and evolution of plant centromeres. Genome Res. 2024, 34, 161–178. [Google Scholar] [CrossRef]
- Scholes, D.T.; Kenny, A.E.; Gamache, E.R.; Mou, Z.M.; Curcio, M.J. Activation of a LTR-retrotransposonby telomere erosion. Proc. Natl. Acad. Sci. USA 2003, 100, 15736–15741. [Google Scholar] [CrossRef]
- Berteli, T.S.; Wang, F.; Kohlrausch, F.B.; Da Luz, C.M.; Oliveira, F.V.; Keefe, D.L.; Navarro, P.A. Impact of superovulation and in vitro fertilization on LINE-1 copy number and telomere length in C57BL/6 J mice blastocysts. Mol. Biol. Rep. 2022, 49, 4909–4917. [Google Scholar] [CrossRef]
- Lu, X.; Liu, L. Genome stability from the perspective of telomere length. Trends Genet. 2024, 40, 175–186. [Google Scholar] [CrossRef]
- Belancio, V.P.; Hedges, D.J.; Deininger, P. LINE-1 RNA splicing and influences on mammalian gene expression. Nucleic Acids Res. 2006, 34, 1512–1521. [Google Scholar] [CrossRef]
- Lev-Maor, G.; Ram, O.; Kim, E.; Sela, N.; Goren, A.; Levanon, E.Y.; Ast, G. Intronic Alus influence alternative splicing. PLoS Genet. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liang, G.; Molloy, P.L.; Jones, P.A. DNA methylation enables transposable element-driven genome expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 19359–19366. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Jiao, B.; Yang, Y.; Shan, L.; Li, T.; Li, X.; Xi, Z.; Wang, X.; Liu, J. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.S.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef]
- Ou, S.; Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef]
- Yan, H.; Bombarely, A.; Li, S. DeepTE: A computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 2020, 36, 4269–4275. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Species a | Total Chromosomes | Genes on Chromosomes | Genome Size (Gb) |
|---|---|---|---|---|
| Cycads | Cycas panzhihuaensis (R) | n = 11 | 31,145 | 10.48 |
| Ginkgophyte | Ginkgo biloba (R) | n = 12 | 27,474 | 9.88 |
| Conifiers | Metasequoia glyptostroboides (R) | n = 11 | 32,013 | 8.07 |
| Sequoiadendron giganteum | n = 11 | 40,016 | 8.13 | |
| Torreya grandis (R) | n = 11 | 46,303 | 19.05 | |
| Taxus yunnanensis | n = 12 | 34,761 | 10.73 | |
| Taxus wallichiana | n = 12 | 44,008 | 11.12 | |
| Pinus tabulaeformis (R) | n = 12 | 76,237 | 25.42 | |
| Gnetophytes | Gnetum montanum | n = 22 | 24,492 | 4.14 |
| Welwitschia mirabilis | n = 21 | 25,593 | 6.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, H.; Zhong, L.; He, Y.; He, J.; Wu, Y.; Guo, Y.; Mei, L.; Wang, G.; Cao, F.; Fu, F.; et al. Construction of Ancestral Chromosomes in Gymnosperms and the Application in Comparative Genomic Analysis. Plants 2025, 14, 2361. https://doi.org/10.3390/plants14152361
Liao H, Zhong L, He Y, He J, Wu Y, Guo Y, Mei L, Wang G, Cao F, Fu F, et al. Construction of Ancestral Chromosomes in Gymnosperms and the Application in Comparative Genomic Analysis. Plants. 2025; 14(15):2361. https://doi.org/10.3390/plants14152361
Chicago/Turabian StyleLiao, Haoran, Lianghui Zhong, Yujie He, Jie He, Yuhan Wu, Ying Guo, Lina Mei, Guibing Wang, Fuliang Cao, Fangfang Fu, and et al. 2025. "Construction of Ancestral Chromosomes in Gymnosperms and the Application in Comparative Genomic Analysis" Plants 14, no. 15: 2361. https://doi.org/10.3390/plants14152361
APA StyleLiao, H., Zhong, L., He, Y., He, J., Wu, Y., Guo, Y., Mei, L., Wang, G., Cao, F., Fu, F., & Xue, L. (2025). Construction of Ancestral Chromosomes in Gymnosperms and the Application in Comparative Genomic Analysis. Plants, 14(15), 2361. https://doi.org/10.3390/plants14152361