
Transcriptome and Endogenous Hormones Reveal the Regulatory Mechanism of Flower Development in Camellia azalea


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Observation and Sampling of C. azalea at Different Developmental Stages
2.2. Statistical and Functional Annotation of Transcriptome Sequencing Data
2.3. C. azalea Sample Correlation Analysis
2.4. DEG Screening and Enrichment Analysis
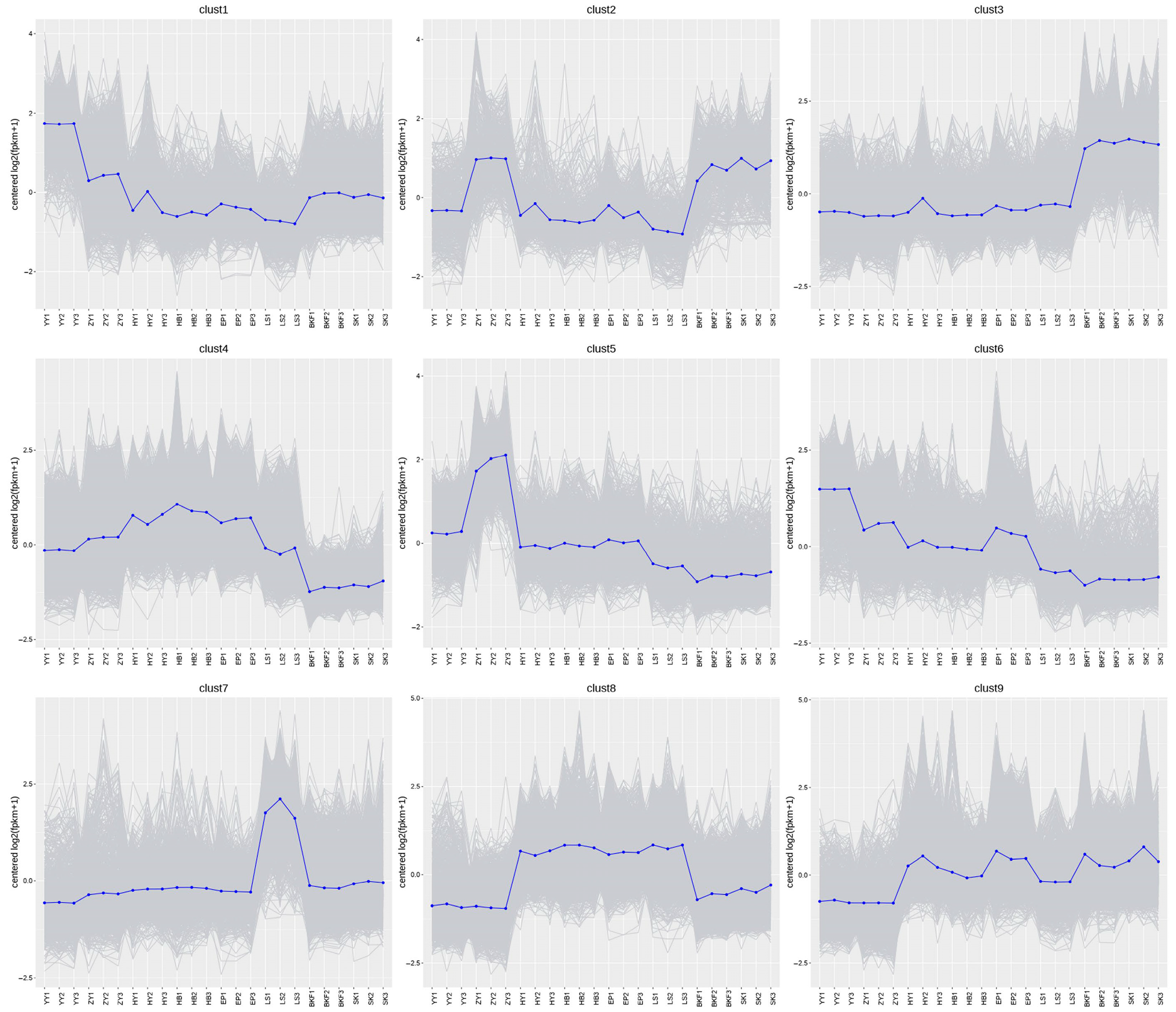
2.5. Cluster Analysis of DEGs During the Flowering Development Process in C. azalea
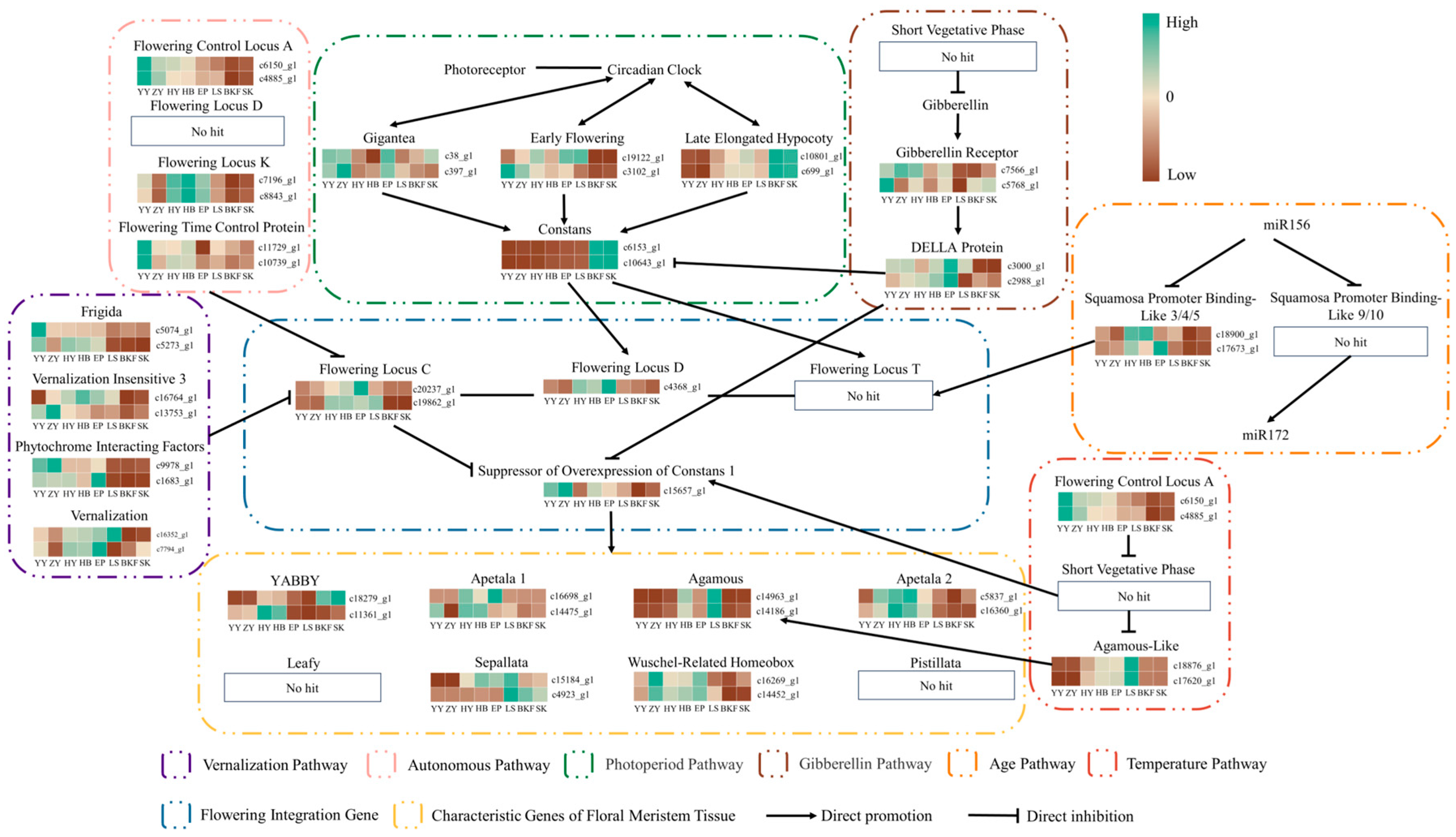
2.6. Expression Pattern Analysis of Key Genes for Flower Formation
2.7. Expression Pattern Analysis of Key Genes Involved in Plant Hormone Signal Transduction
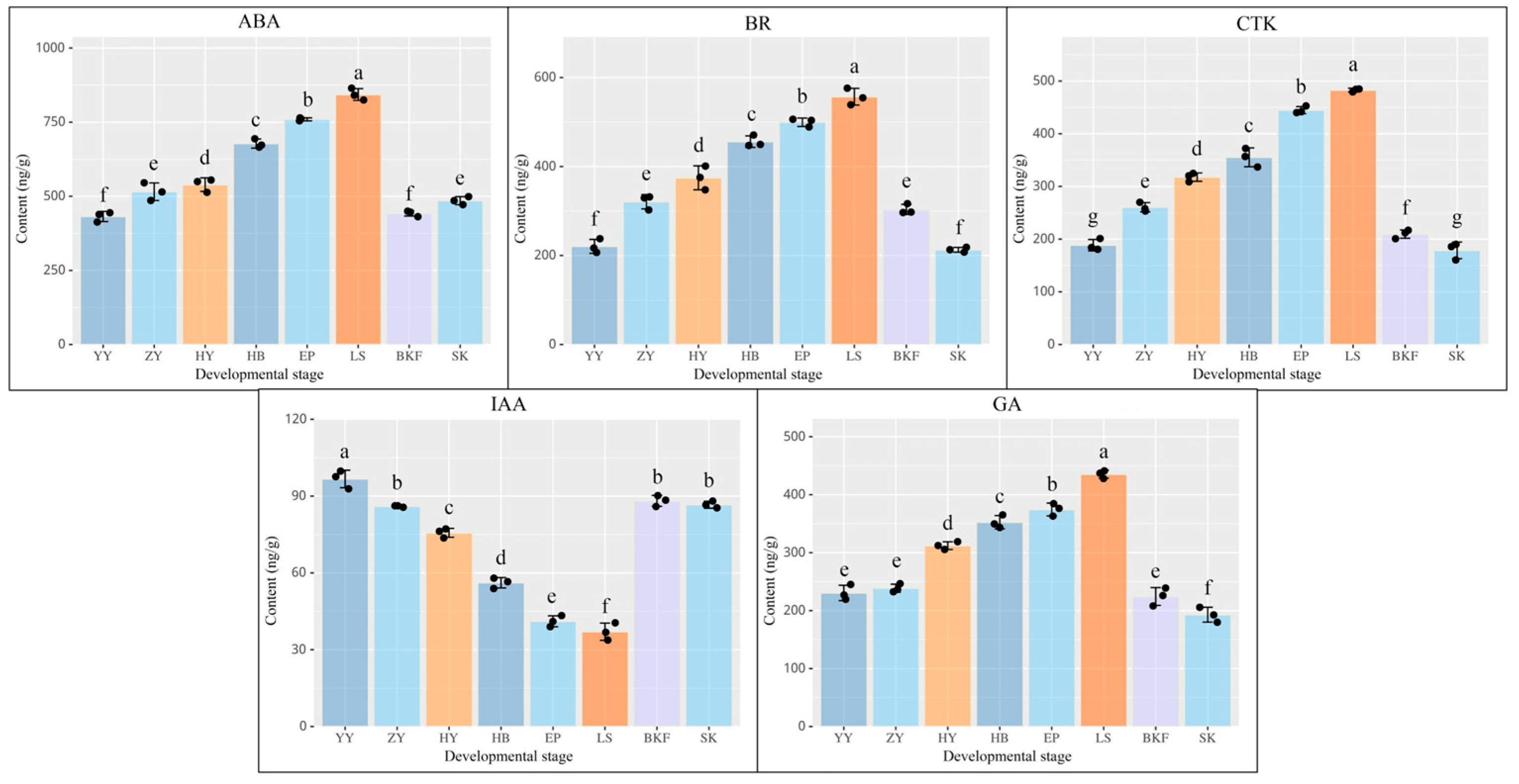
2.8. Determination of Endogenous Hormone Content in C. azalea During Development
2.9. Analysis of Endogenous Hormones and Key Gene Correlations
2.10. qRT-PCR Validation
3. Discussion
3.1. Sample Correlation and PCA
3.2. Identification and Functional Annotation of DEGs at Different Growth Stages of C. azalea
3.3. The Role of Various Floral Pathways in the Development of the Flower of C. azalea
3.4. Effects of Plant Hormones on Flower Formation in C. azalea
3.5. Comparison of Flowering Mechanisms Between C. azalea and Most Camellia Species
4. Materials and Methods
4.1. Collection of Sample Materials
4.2. Determination of Endogenous Hormone Content
4.3. RNA Extraction and Detection
4.4. Library Construction and Sequencing
4.5. Differential Expressed Genes (DEGs) Screening and Enrichment Analysis
4.6. Real-Time PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Boss, P.K. Multiple pathways in the decision to flower: Enabling promoting and resetting. Plant Cell 2004, 16 (Suppl. S1), S18–S31. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.A.; Bailey, P.C.; Laurie, D.A. Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS ONE 2010, 5, e10065. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Takahashi, N.; Hirata, Y.; Ronald, J.; Porco, S.; Davis, S.; Nusinow, D.A.; Kay, S.; Mas, P. A mobile ELF4 delivers circadian temperature information from shoots to roots. Nat. Plants 2020, 6, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Ito, S.; Imaizumi, T. Flowering time regulation: Photoperiod and temperature-sensing inleaves. Trends Plant Sci. 2013, 18, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Lu, S.; Jiang, J.H.; Li, S.R.; Zhang, N.; Jiang, X.Y.; Wu, F. Research Progress on Influencing Factors and Mechanisms of Flower Bud Differentiation in Horticultural Plants. Acta Hortic. Sin. 2023, 50, 1151–1164. [Google Scholar] [CrossRef]
- Fornara, F.; Montaigu, A.D.; Coupland, G. Snap Shot: Control of flowering in Arabidopsis. Cell 2010, 141, 550. [Google Scholar] [CrossRef] [PubMed]
- Valverde, F.; Mouradov, A.; Soppe, W.; Ravenscroft, D.; Samach, A.; Coupland, G. Photoreceptor Regulation of CONSTANS Protein in Photoperiodic Flowering. Science 2004, 303, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Viswanath, K.K.; Tong, C.G.; An, H.R.; Chen, F.C. Floral induction and flower development of orchids. Front. Plant Sci. 2019, 10, 1258. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.R.; Shu, W.B.; Qiu, M.X.; Li, L.Y.; Han, L.; Sun, Z.Y.; Ju, G.S.; Li, Z.J. Current progress on gene regulation in the floral organ and flower formation of orchid. J. China Agric. Univ. 2023, 28, 57–67. [Google Scholar]
- Chen, S.; Wang, P.; Kong, W.; Chai, K.; Zhang, S.; Yu, J.; Wang, Y.; Jiang, M.; Lei, W.; Chen, X. Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia sinensis. Nat. Plants 2023, 9, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.Q.; Li, J.Y.; Li, X.L.; Wu, B.; Wang, J.Y.; Liu, Z.C.; Yin, H.F. Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia azalea. Sci. Rep. 2015, 5, 9729. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Zhu, P.B.; Ge, J.T.; Hui, L.C.; Li, J.Y.; Sun, M.W.; Zhao, T.L.; Shao, X.B.; Tang, X.Y. Cloning and Expression Analysis of ACO1 from Camellia azalea. Chin. J. Trop. Crops 2021, 42, 2175–2182. [Google Scholar] [CrossRef]
- Cui, J. Molecular Mechanism of Floral Induction Difference Between C. perpetua and C. petelotii. Master’s Thesis, Guangxi University, Nanning, China, 2022. [Google Scholar] [CrossRef]
- Zou, L.P.; Pan, C.; Wang, M.X.; Cui, L.; Han, B.Y. Progress on the mechanism of hormones regulating plant flower Formation. Hereditas 2020, 42, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhang, S.S.; Bao, M.Z.; Liu, G.F. Advances on Molecular Mechanism of Floral Initiation in Higher Plants. Mol. Plant Breed. 2018, 16, 3681–3692. [Google Scholar] [CrossRef]
- Wilson, R.N.; Heckman, J.W.; Somerville, C.R. Gibberellin is required for flowering in Arabidopsis thaliana under short days. Plant Physiol. 1992, 100, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.P. The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr. Biol. 2011, 21, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, D.C.; Villalobos, L.; Abel, S. Structural Biology of Nuclear Auxin Action. Trends Plant 2016, 21, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Zhao, J.Q.; Wang, X.T.; Shi, X.B.; Wang, B.L.; Zheng, X.C.; Liu, F.Z. The Influence of Changes of Endogenous Hormones in Shoot on the Grapes Flower Bud Differentiation in Greenhouse. Sci. Agric. Sin. 2014, 47, 4695–4705. [Google Scholar] [CrossRef]
- Jiang, H.D.; Sun, F.F.; Qin, H.Z.; Tang, J.M.; Wei, X.; Chai, S.F. Flower bud differentiation and leaf endogenous hormone changes of Camellia perpetua. Guihaia 2024, 44, 56–67. [Google Scholar] [CrossRef]
- Oneill, D.P.; Ross, J. Auxin Regulation of the Gibberellin Pathway in Pea. Plant Physiol. 2003, 130, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Matteo, R.; Alice, R.T.; Massimo, G.; Chiara, T.; Lucio, C. ABA-dependent control of GIGANTEA signalling enables drought escape via up-regulation of FLOWERING LOCUS T in Arabidopsis thaliana. J. Exp. Bot. 2016, 22, 6309–6322. [Google Scholar] [CrossRef]
- D’Aloia, M.; Bonhomme, D.; Bouche, F.; Tamseddak, K.; Ormenese, S.; Torti, S.; Coupland, G.; Perilleux, C. Cytokinin promotes flowering of Arabidopsis via transcriptional activation of the FT paralogue TSF. Plant J. 2011, 65, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Ni, M.; Zhang, Z.H.; Yu, F.Y. Research progress of brassinolide in regulating plant growth and development. Sci. Silvae Sin. 2022, 58, 144–155. [Google Scholar]
- Wang, R.Q.; Xu, Y.; Zhang, M.; Hao, G.; Zhao, Q.M.; Liu, X.K.; Liu, X.F.; Yu, B.; Zhang, W.J. Genetic structure and demographic analysis of a true single-population species, Camellia azalea. BMC Plant Biol. 2024, 24, 1272. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Huang, Z.W.; Lu, J.S.; Cui, X.Q.; Su, Q.; Li, C.N.; Bu, C.Y. Physiological Study on Grafted Affinity of Camellia azalea with Different Species Bud Seedling Rootstocks. Chin. J. Trop. Crops 2022, 43, 978–985. [Google Scholar] [CrossRef]
- Li, X.M.; Li, C.N.; Liu, X.L.; Bu, C.Y.; Huang, Z.W. Effect of Shading on Leaf Growth and Primary Metabolism of Camellia azalea Seedlings. Acta Bot. Boreali-Occident. Sin. 2019, 39, 294–301. [Google Scholar] [CrossRef]
- Zhu, G.P.; Li, J.Y. Research Progress of Rare and Endangered Camellia changii Ye. Subtrop. Plant Sci. 2009, 38, 83–89. [Google Scholar] [CrossRef]
- Li, X.L.; Sun, Z.Y.; Li, J.Y.; Fan, Z.Q.; Yin, H.F. Changes of Metabolites and Flower Bud Differentiation of Camellia azalea. Sci. Silvae Sin. 2012, 48, 81–86. [Google Scholar] [CrossRef]
- Jang, S.; Hur, J.; Kim, S.J.; Han, M.J.; Kim, S.R.; An, G. Ectopic expression of OsYAB1 causes extra stamens and carpels in rice. Plant Mol. Biol. 2004, 56, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Stahle, M.I.; Kuehlich, J.; Staron, L.; Arnim, A.; Golz, J.F. YABBYs and the transcriptional corepressors LEUNIG and LEUNIG_HOMOLOG maintain leaf polarity and meristem activity in Arabidopsis. Plant Cell 2009, 21, 3105–3118. [Google Scholar] [CrossRef] [PubMed]
- Boss, P.K.; Sensi, E.; Hua, C.; Thomas, M.R. Cloning and characterisation of grapevine (Vitis vinifera L.) MADS-box genes expressed during inflorescence and berry development. Plant Sci. 2002, 162, 887–895. [Google Scholar] [CrossRef]
- Feng, S.S.; Wang, L.; Zhou, Y.; Wang, Y.P.; Fang, Y.J. Research Progresses on WOX Family Genes in Regulating Plant Development and Abiotic Stress Response. Biotechnol. Bull. 2023, 39, 1–13. [Google Scholar] [CrossRef]
- Du, M.; Spalding, E.P.; Gray, W.M. Rapid auxin-mediated cell expansion. Annu. Rev. Plant Biol. 2020, 71, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gao, S.Z.; Lu, H.; Zhan, Y.G.; Zeng, F.S. Plant Cell Wall Development and Its Function in Abiotic Stress. Plant Physiol. J. 2023, 59, 1251–1264. [Google Scholar] [CrossRef]
- Brumos, J.; Alonso, J.M.; Stepanova, A.N. Genetic aspects of auxin biosynthesis and its regulation. Physiol. Plant. 2014, 151, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zeng, Y.L.; Zhang, F.C. The Regulation of Trp-Asp Repeat Sequence Protein (WD40) in Plants Response to Stress. Plant Physiol. J. 2013, 49, 1160–1167. [Google Scholar] [CrossRef]
- Qi, Z.Y.; Wang, T.; Sang, K.Q.; Liu, Y.; Wang, M.Q.; Yu, J.Q.; Zhou, Y.H.; Xia, X.J. Effects of Supplemental Lighting at Different Positions on Tomato Plant Morphology, Photosynthesis and Endogenous Hormone Biosynthesis Under Low-light Environment. Hortic. Sin. 2021, 48, 1504–1516. [Google Scholar] [CrossRef]
- Zhao, L.Q.; Zhang, X.L.; Li, F.; Tan, Y.; Shi, W.L.; Huang, M.J.; Huang, H.Q. Cloning and expression analysis of ImLHY and ImC4H genes in Impatiens morsei Hook. F. J. South. Agric. 2023, 54, 3464–3474. [Google Scholar] [CrossRef]
- Shim, J.S.; Kubota, A.; Imaizumi, T. Circadian clock and photoperiodic flowering in Arabidopsis: CONSTANS isa hub for signal integration. Plant Physiol. 2017, 173, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Suárez-López, P.; Wheatley, K.; Robson, F.; Onouchi, H.; Valverde, F.; Coupland, G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dou, L.D.; Zhang, H.J. Molecular and Genetic Mechanisms of Control of Floral Induction in Higher Plants. Plant Physiol. J. 2014, 50, 1459–1468. [Google Scholar] [CrossRef]
- Deng, W.W.; Ying, H.; Helliwell, C.A.; Taylor, J.M.; Peacock, W.J.; Dennis, E.S. FLOWERING LOCUS C (FLC) regulates development pathways throughout the life cycle of Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 6680–6685. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Seo, P.J.; Ahn, J.H.; Park, C.M. Arabidopsis RNA-binding Protein FCA Regulates MicroRNA172 Processing in Thermosensory Flowering. J. Biol. Chem. 2012, 287, 16007–16016. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.X.; Chen, R.; Shi, Y.; Gai, C.Y.; Fan, K.; Li, Z.W. Research progress on the biological functions of SPL transcription factors in plants. Chin. Bull. Bot. 2023, 58, 982–997. [Google Scholar] [CrossRef]
- Xu, M.L.; Hu, T.Q.; Zhao, J.F.; Park, M.Y.; Earley, K.W.; Wu, G.; Yang, L.; Poethig, R.S. Developmental Functions of miR156-Regulated SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) Genes in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006263. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Willige, B.C. Shedding light on gibberellic acid signaling. Curr. Opin. Plant Biol. 2009, 12, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Galvao, V.C.; Zhang, Y.C.; Horrer, D.; Zhang, T.Q.; Hao, Y.H.; Feng, Y.Q.; Wang, S.; Schmid, M.; Wang, J.W. Gibberellin Regulates the Arabidopsis Floral Transition through miR156-Targeted SQUAMOSA PROMOTER BINDING–LIKE Transcription Factors. Plant Cell 2012, 24, 3320–3332. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.; Murase, K.; Rieu, I.; Zentella, R.; Thomas, S.G. Genetic characterization and functional analysis of the GID1 gibberellin receptors in Arabidopsis. Plant Cell 2006, 18, 3399–3414. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Lei, C.; Xin, Z.M.; Shao, H.X.; Muhammad, M.T.; Zhang, D.; Han, C.Y. Cloning and Expression Analysis of the Gibberellin Signal Transduction Factor MdGAMYB in Malus. Acta Hortic. Sin. 2017, 44, 817–827. [Google Scholar] [CrossRef]
- Li, S.B.; Xie, Z.Z.; Hu, C.G.; Zhang, J.Z. A review of auxin response factors(ARFs)in Plants. Front. Plant Sci. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhou, J.; Dang, S.Z.; Tang, X.S.; Zhang, Y.H. Functional Analysis of VvARF18 Gene in Red Globe Grape. Sci. Agric. Sin. 2024, 57, 1363–1376. [Google Scholar] [CrossRef]
- Zhou, Y.; Mo, Y.X.; Liu, S.Y.; Li, W.J.; Li, H.L.; Liu, K.D. Transcriptome-wide Identification of the ARF Gene Family in Sugar Apple (Annona squamosa L.) and Expression Analysis During Flower Development. Chin. J. Trop. Crops 2023, 44, 661–672. [Google Scholar] [CrossRef]
- Feng, H.Q.; Li, C. Research Advances of Auxin Signal Transduction. Biotechnol. Bull. 2018, 34, 24–30. [Google Scholar] [CrossRef]
- Li, H.F.; Zhang, W.Q.; Dong, Q.L.; Wang, X.F.; Ran, K. Cloning, sequencing and expression analysis of auxin response factors (MdARF) in apple. J. Fruit Sci. 2018, 35, 1170–1181. [Google Scholar] [CrossRef]
- Salehin, M.; Bagchi, R.; Estelle, M. SCFTIR1/AFB-Based Auxin Perception: Mechanism and Role in Plant Growth and Development. Plant Cell 2015, 27, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Annie, J.; Isabelle, G.; Georges, B. Cell division and morphological changes in the shoot apex of Arabidopsis thaliana during floral transition. Ann. Bot. 2003, 5, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Bernier, G.; Corbesier, L.; Périlleux, C. The flowering process: On the track of controlling factors in Sinapis alba. Russ. J. Plant Physiol. 2002, 49, 445–450. [Google Scholar] [CrossRef]
- Liu, Y.; Dreni, L.; Zhang, H.J.; Zhang, X.Z.; Li, N.; Zhang, K.X.; Di, T.M.; Wang, L.; Yang, Y.J.; Hao, X.Y.; et al. A Tea Plant (Camellia sinensis) FLOWERING LOCUS C-like Gene, CsFLC1, Is Correlated to Bud Dormancy and Triggers Early Flowering in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 15711. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Guo, T.; Zou, S.; Li, L.; Li, X.; Wang, J.; Zhu, Z.; Ai, L. Integrative Transcriptomic and Metabolomic Analysis Reveals the Molecular Mechanisms Underlying Flowering Time Variation in Camellia Species. Agronomy 2025, 15, 1288. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, X.Y.; Zheng, M.X.; Wang, X.C.; Xiao, B.; Yang, Y.J. Recent advances on Tea Flowering Mechanisms. J. Tea Sci. 2019, 39, 1–10. [Google Scholar] [CrossRef]
- Li, Y.L. Study on the Effects of Different Photoperiod on the Inflorescence of Camellia japonica. Master’s Thesis, Sichuan Agricultural University, Chengdu, China, 2005. [Google Scholar]
- Wang, J.Y.; Wu, B.; Yin, H.F.; Fan, Z.Q.; Li, X.L.; Ni, S.; He, L.B.; Li, J.Y. Overexpression of CaAPX Induces Orchestrated Reactive Oxygen Scavenging and Enhances Cold and Heat Tolerances in Tobacco. BioMed Res. Int. 2017, 354, 1–15. [Google Scholar] [CrossRef]
- Blazquez, M.A.; Ahn, J.H.; Weigel, D. A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nat. Genet. 2003, 33, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Identification of Flowering-Related Genes and Function Analysis of CsFLC1 Gene in Tea Plants. Ph.D. Thesis, Northwest A&F University, Xianyang, China, 2022. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Yang, F.; Nie, R.; Zhao, W.; Geng, F.; Chen, L. Transcriptome and Endogenous Hormones Reveal the Regulatory Mechanism of Flower Development in Camellia azalea. Plants 2025, 14, 2291. https://doi.org/10.3390/plants14152291
Xu J, Yang F, Nie R, Zhao W, Geng F, Chen L. Transcriptome and Endogenous Hormones Reveal the Regulatory Mechanism of Flower Development in Camellia azalea. Plants. 2025; 14(15):2291. https://doi.org/10.3390/plants14152291
Chicago/Turabian StyleXu, Jian, Fan Yang, Ruimin Nie, Wanyue Zhao, Fang Geng, and Longqing Chen. 2025. "Transcriptome and Endogenous Hormones Reveal the Regulatory Mechanism of Flower Development in Camellia azalea" Plants 14, no. 15: 2291. https://doi.org/10.3390/plants14152291
APA StyleXu, J., Yang, F., Nie, R., Zhao, W., Geng, F., & Chen, L. (2025). Transcriptome and Endogenous Hormones Reveal the Regulatory Mechanism of Flower Development in Camellia azalea. Plants, 14(15), 2291. https://doi.org/10.3390/plants14152291