
Genebank Management Through Microsatellite Markers: A Case Study in Two Italian Peach Germplasm Collections
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Genetic Diversity Analysis and Collection Characterization
2.2. Parentage Analysis
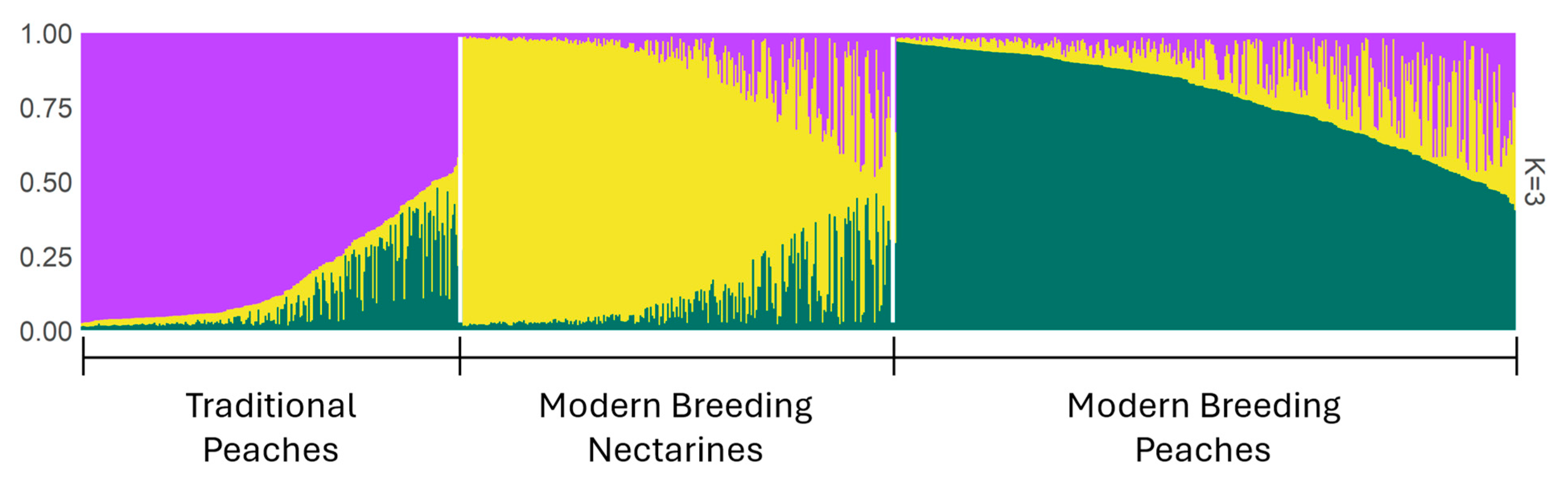
2.3. Population Structure
3. Materials and Methods
3.1. Plant Materials and DNA Extraction
3.2. SSR Analysis
3.3. Genetic Diversity Analysis
3.4. Population Structure Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NGFC CREA | National Fruit Germplasm Collection |
| MAS.PES | Breeding Apricot and Peach through Marker-Assisted Selection |
| ITPGRFA | International Treaty for Plant Genetic Resources for Food and Agriculture |
| GWAS | Genome-Wide Association Studies |
| GBS | Genotyping by Sequencing |
| SSRs | Simple Sequence Repeats |
| SNPs | Single Nucleotide Polymorphisms |
| PD | Discrimination Power |
| Ho | Observed Heterozygosity |
| He | Expected Heterozygosity |
| PIC | Polymorphic Information Content |
| PI | Probability of Identity |
| MAF | Minor Allele Frequency |
References
- Potter, D.; Eriksson, T.; Evans, R.C.; Oh, S.; Smedmark, J.E.E.; Morgan, D.R.; Kerr, M.; Robertson, K.R.; Arsenault, M.; Dickinson, T.A.; et al. Phylogeny and Classification of Rosaceae. Plant Syst. Evol. 2007, 266, 5–43. [Google Scholar] [CrossRef]
- Lee, S.; Wen, J. A Phylogenetic Analysis of Prunus and the Amygdaloideae (Rosaceae) Using ITS Sequences of Nuclear Ribosomal DNA. Am. J. Bot. 2001, 88, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V.; Korban, S.S.; Sosinski, B.; Abbott, A.G.; Aldwinckle, H.S.; Folta, K.M.; Iezzoni, A.; Main, D.; Arús, P.; Dandekar, A.M.; et al. Multiple Models for Rosaceae Genomics. Plant Physiol. 2008, 147, 985–1003. [Google Scholar] [CrossRef] [PubMed]
- Boudehri, K.; Bendahmane, A.; Cardinet, G.; Troadec, C.; Moing, A.; Dirlewanger, E. Phenotypic and Fine Genetic Characterization of the D Locus Controlling Fruit Acidity in Peach. BMC Plant Biol. 2009, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Gilani, S.A.; Qureshi, R.A.; Khan, A.M.; Potter, D. A Molecular Phylogeny of Selected Species of Genus Prunus L. (Rosaceae) from Pakistan Using the Internal Transcribed Spacer (ITS) Spacer DNA. Afr. J. Biotechnol. 2010, 9, 4867–4872. [Google Scholar]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The High-Quality Draft Genome of Peach (Prunus persica) Identifies Unique Patterns of Genetic Diversity, Domestication and Genome Evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef]
- Faust, M.; Timon, B. Origin and Dissemination of Peach. Hortic. Rev. 1995, 17, 331–379. [Google Scholar] [CrossRef]
- Vaccino, P.; Antonetti, M.; Balconi, C.; Brandolini, A.; Cappellozza, S.; Caputo, A.R.; Carboni, A.; Caruso, M.; Copetta, A.; de Dato, G.; et al. Plant Genetic Resources for Food and Agriculture: The Role and Contribution of CREA (Italy) within the National Program RGV-FAO. Agronomy 2024, 14, 1263. [Google Scholar] [CrossRef]
- Van Treuren, R.; Van Hintum, T.J.L. Marker-Assisted Reduction of Redundancy in Germplasm Collections: Genetic and Economic Aspects. Acta Hortic. 2003, 623, 139–149. [Google Scholar] [CrossRef]
- Bettoni, J.C.; Bonnart, R.; Volk, G.M. Challenges in Implementing Plant Shoot Tip Cryopreservation Technologies. Plant Cell Tissue Organ Cult. 2021, 144, 21–34. [Google Scholar] [CrossRef]
- Tripodi, P.; Rabanus-Wallace, M.T.; Barchi, L.; Kale, S.; Esposito, S.; Acquadro, A.; Schafleitner, R.; van Zonneveld, M.; Prohens, J.; Diez, M.J.; et al. Global Range Expansion History of Pepper (Capsicum spp.) Revealed by over 10,000 Genebank Accessions. Proc. Natl. Acad. Sci. USA 2021, 118, e2104315118. [Google Scholar] [CrossRef]
- Emanuelli, F.; Lorenzi, S.; Grzeskowiak, L.; Catalano, V.; Stefanini, M.; Troggio, M.; Myles, S.; Martinez-Zapater, J.M.; Zyprian, E.; Moreira, F.M.; et al. Genetic Diversity and Population Structure Assessed by SSR and SNP Markers in a Large Germplasm Collection of Grape. BMC Plant Biol. 2013, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- van Treuren, R.; Kemp, H.; Ernsting, G.; Jongejans, B.; Houtman, H.; Visser, L. Microsatellite Genotyping of Apple (Malus × domestica Borkh.) Genetic Resources in the Netherlands: Application in Collection Management and Variety Identification. Genet. Resour. Crop. Evol. 2010, 57, 853–865. [Google Scholar] [CrossRef]
- Liang, W.; Dondini, L.; De Franceschi, P.; Paris, R.; Sansavini, S.; Tartarini, S. Genetic Diversity, Population Structure and Construction of a Core Collection of Apple Cultivars from Italian Germplasm. Plant Mol. Biol. Rep. 2015, 33, 458–473. [Google Scholar] [CrossRef]
- Lassois, L.; Denancé, C.; Ravon, E.; Guyader, A.; Guisnel, R.; Hibrand-Saint-Oyant, L.; Poncet, C.; Lasserre-Zuber, P.; Feugey, L.; Durel, C.E. Genetic Diversity, Population Structure, Parentage Analysis, and Construction of Core Collections in the French Apple Germplasm Based on SSR Markers. Plant Mol. Biol. Rep. 2016, 34, 827–844. [Google Scholar] [CrossRef]
- Larsen, B.; Toldam-Andersen, T.B.; Pedersen, C.; Ørgaard, M. Unravelling Genetic Diversity and Cultivar Parentage in the Danish Apple Gene Bank Collection. Tree Genet. Genomes 2017, 13, 14. [Google Scholar] [CrossRef]
- Pina, A.; Urrestarazu, J.; Errea, P. Analysis of the Genetic Diversity of Local Apple Cultivars from Mountainous Areas from Aragon (Northeastern Spain). Sci. Hortic. 2014, 174, 1–9. [Google Scholar] [CrossRef]
- Denancé, C.; Muranty, H.; Durel, C.E. MUNQ—Malus UNiQue Genotype Code for Grouping Apple Accessions Corresponding to a Unique Genotypic Profile; INRAE: Paris, France, 2020. [Google Scholar]
- Larsen, B.; Howard, N.P.; Denancé, C.; Durel, C.E.; Pedersen, C.; Sätra, J.S.; Garkava-Gustavsson, L.; Troggio, M.; van de Weg, E. Cultivar Fingerprinting and SNP-Based Pedigree Reconstruction in Danish Heritage Apple Cultivars Utilizing Genotypic Data from Multiple Germplasm Collections in the World. Genet. Resour. Crop. Evol. 2024, 72, 2397–2411. [Google Scholar] [CrossRef]
- Aranzana, M.J.; Carbó, J.; Arús, P. Microsatellite Variability in Peach [Prunus persica (L.) Batsch]: Cultivar Identification, Marker Mutation, Pedigree Inferences and Population Structure. Theor. Appl. Genet. 2003, 106, 1341–1352. [Google Scholar] [CrossRef]
- Aranzana, M.J.; Abbassi, E.K.; Howad, W.; Arús, P. Genetic Variation, Population Structure and Linkage Disequilibrium in Peach Commercial Varieties. BMC Genet 2010, 11, 69. [Google Scholar] [CrossRef]
- Li, X.W.; Meng, X.Q.; Jia, H.J.; Yu, M.L.; Ma, R.J.; Wang, L.R.; Cao, K.; Shen, Z.J.; Niu, L.; Tian, J.B.; et al. Peach Genetic Resources: Diversity, Population Structure and Linkage Disequilibrium. BMC Genet. 2013, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Méndez, M.; Rojas, G.; Muñoz, C.; Lemus, G.; Hinrichsen, P. Identification of a Minimal Microsatellite Marker Panel for the Fingerprinting of Peach and Nectarine Cultivars; Universidad de Chile: Santiago, Chile, 2008. [Google Scholar]
- Bouhadida, M.; Moreno, M.Á.; Gonzalo, M.J.; Alonso, J.M.; Gogorcena, Y. Genetic Variability of Introduced and Local Spanish Peach Cultivars Determined by SSR Markers. Tree Genet. Genomes 2011, 7, 257–270. [Google Scholar] [CrossRef]
- Chavez, D.J.; Beckman, T.G.; Werner, D.J.; Chaparro, J.X. Genetic Diversity in Peach [Prunus Persica (l.) Batsch] at the University of Florida: Past, Present and Future. Tree Genet. Genomes 2014, 10, 1399–1417. [Google Scholar] [CrossRef]
- Shen, Z.J.; Ma, R.J.; Cai, Z.X.; Yu, M.L. Diversity, Population Structure, And Evolution of Local Peach Cultivars in China Identified by Simple Sequence Repeats. Genet. Mol. Res. 2015, 14, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Micheletti, D.; Dettori, M.T.; Micali, S.; Aramini, V.; Pacheco, I.; Da Silva Linge, C.; Foschi, S.; Banchi, E.; Barreneche, T.; Quilot-Turion, B.; et al. Whole-Genome Analysis of Diversity and SNP-Major Gene Association in Peach Germplasm. PLoS ONE 2015, 10, e0136803. [Google Scholar] [CrossRef]
- Verde, I.; Bassil, N.; Scalabrin, S.; Gilmore, B.; Lawley, C.T.; Gasic, K.; Micheletti, D.; Rosyara, U.R.; Cattonaro, F.; Vendramin, E.; et al. Development and Evaluation of a 9K SNP Array for Peach by Internationally Coordinated SNP Detection and Validation in Breeding Germplasm. PLoS ONE 2012, 7, e35668. [Google Scholar] [CrossRef]
- Cao, K.; Zhou, Z.; Wang, Q.; Guo, J.; Zhao, P.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wang, X.; et al. Genome-Wide Association Study of 12 Agronomic Traits in Peach. Nat. Commun. 2016, 7, 13246. [Google Scholar] [CrossRef]
- Guo, J.; Cao, K.; Deng, C.; Li, Y.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wu, J.; Guan, L.; et al. An Integrated Peach Genome Structural Variation Map Uncovers Genes Associated with Fruit Traits. Genome Biol. 2020, 21, 258. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, K.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Zhao, P.; Guo, J.; Ding, T.; Guan, L.; et al. Genomic Analyses of an Extensive Collection of Wild and Cultivated Accessions Provide New Insights into Peach Breeding History. Genome Biol. 2019, 20, 36. [Google Scholar] [CrossRef]
- Yu, Y.; Guan, J.; Xu, Y.; Ren, F.; Zhang, Z.; Yan, J.; Fu, J.; Guo, J.; Shen, Z.; Zhao, J.; et al. Population-Scale Peach Genome Analyses Unravel Selection Patterns and Biochemical Basis Underlying Fruit Flavor. Nat. Commun. 2021, 12, 3604. [Google Scholar] [CrossRef]
- Bassi, D.; Foschi, S.; Rizzo, M.; Castellari, L. Progetto ‘MAS.PES‘ per Il Miglioramento Genetico Del Pesco: Criteri Di Selezione e Individuazione Ideotipi Di Riferimento. Italus Hortus 2010, 17, 60–62. [Google Scholar]
- Paula LA, B.V.F.J. Caracterização Molecular Variabilidade Genética Entre Porta-Enxertos de Pessegueiro Com Base Em Marcadores Codominantes. Pesqui. Agropecu. Bras. Brasília 2012, 47, 193–199. [Google Scholar] [CrossRef]
- Chalak, L.; Chehade, A.; Elbitar, A.; Cosson, P.; Zanetto, A.; Dirlewanger, E.; Laigret, F. Morphological and Molecular Characterization of Peach Accessions (Prunus persica L.) Cultivated in Lebanon. Lebanese Sci. J. 2003, 7, 23–31. [Google Scholar]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a Genetic Linkage Map in Man Using Restriction Fragment Length Polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar] [PubMed]
- Giovannini, D.; Dettori, M.T.; Leone, A.; Liverani, A.; Sirri, S.; Tellarini, S.; Verde, I. Assessment of Genetic Variability in Italian Heritage Peach Resources from Emilia-Romagna Using Microsatellite Markers. J. Hortic. Sci. Biotechnol. 2012, 87, 435–440. [Google Scholar] [CrossRef]
- Falchi, R.; Vendramin, E.; Zanon, L.; Scalabrin, S.; Cipriani, G.; Verde, I.; Vizzotto, G.; Morgante, M. Three Distinct Mutational Mechanisms Acting on a Single Gene Underpin the Origin of Yellow Flesh in Peach. Plant J. 2013, 76, 175–187. [Google Scholar] [CrossRef]
- Adami, M.; Franceschi, P.; Brandi, F.; Liverani, A.; Giovannini, D.; Rosati, C.; Dondini, L.; Tartarini, S. Identifying a Carotenoid Cleavage Dioxygenase (Ccd4) Gene Controlling Yellow/White Fruit Flesh Color of Peach. Plant Mol. Biol. Rep. 2013, 31, 1166–1175. [Google Scholar] [CrossRef]
- Cao, K.; Zheng, Z.; Wang, L.; Liu, X.; Zhu, G.; Fang, W.; Cheng, S.; Zeng, P.; Chen, C.; Wang, X.; et al. Comparative Population Genomics Reveals the Domestication History of the Peach, Prunus persica, and Human Influences on Perennial Fruit Crops. Genome Biol. 2014, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Smykov, A.; Fedorova, O.; Shishova, T.; Ivashchenko, I. Introduction and Use of the Peach Gene Pool from China in Nikita Botanical Garden. Acta Hortic. 2018, 1208, 1–6. [Google Scholar] [CrossRef]
- Dabov, S. Inheritance of Powdery Mildew Resistance in the Peach. IV. Data Supporting the Hypothesis about the Main Role of 2 Loci Controlling the Reaction to the Pathogen. Genet. Sel. 1983, 16, 349–355. [Google Scholar]
- Testolin, R.; Marrazzo, T.; Cipriani, G.; Quarta, R.; Verde, I.; Dettori, M.T.; Pancaldi, M.; Sansavini, S. Microsatellite DNA in Peach (Prunus persica L. Batsch) and Its Use in Fingerprinting and Testing the Genetic Origin of Cultivars. Genome 2011, 43, 512–520. [Google Scholar] [CrossRef]
- Okie, W.R. Handbook of Peach and Nectarine Varieties; National Technica; National Technical Information Service, distributor; U.S. Department of Agriculture, Agricultural Research Service: Springfild, VA, USA, 1998.
- Vendramin, E.; Pea, G.; Dondini, L.; Pacheco, I.; Dettori, M.T.; Gazza, L.; Scalabrin, S.; Strozzi, F.; Tartarini, S.; Bassi, D.; et al. A Unique Mutation in a MYB Gene Cosegregates with the Nectarine Phenotype in Peach. PLoS ONE 2014, 9, e90574. [Google Scholar] [CrossRef]
- Scorza, R.; Mehlenbacher, S.A.; Lightner, G.W. Inbreeding and Coancestry of Freestone Peach Cultivars of the Eastern United States and Implications for Peach Germplasm Improvement. J. Am. Soc. Hortic. Sci. 1985, 110, 547–552. [Google Scholar] [CrossRef]
- Vendramin, E.; Linge, C.D.; Bassi, D.; Micali, S.; Chietera, G.; Dettori, M.T.; Verde, I. Atlante Dei Fruttiferi Autoctoni Italiani, 2016th ed.; Carlo, F., Ed.; CREA Olivicoltura, Frutticoltura e Agrumicoltura & MIPAAF: Rome, Italy, 2016; Volume I–III.
- Jouy, C.; Gandelin, M.H.; Guitouni, C.; Pascal, T.; Tuero, C.; Zubor, L.; Badenes, M.T.; Soler, G.; Martinez, J.; Verde, I.; et al. Management of Peach Tree Reference Collections: Ongoing Research & Development Program Relevant to the Community Plant Variety Rights Protection System. Acta Hortic. 2012, 962, 51–56. [Google Scholar] [CrossRef]
- Hayden, M.J.; Nguyen, T.M.; Waterman, A.; Chalmers, K.J. Multiplex-Ready PCR: A New Method for Multiplexed SSR and SNP Genotyping. BMC Genom. 2008, 9, 80. [Google Scholar] [CrossRef]
- Eduardo, I.; Pacheco, I.; Chietera, G.; Bassi, D.; Pozzi, C.; Vecchietti, A.; Rossini, L. QTL Analysis of Fruit Quality Traits in Two Peach Intraspecific Populations and Importance of Maturity Date Pleiotropic Effect. Tree Genet. Genomes 2011, 7, 323–335. [Google Scholar] [CrossRef]
- Pacheco, I.; Bassi, D.; Eduardo, I.; Ciacciulli, A.; Pirona, R.; Rossini, L.; Vecchietti, A. QTL mapping for brown rot (Monilinia fructigena) resistance in an intraspecific peach (Prunus persica L. Batsch) F1 progeny. Tree Genet. Genomes 2014, 10, 1223–1242. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-Pc, Numerical Taxonomy and Multivariate Analysis System (V. 2.11Q); Applied Biostatistics: Port Jefferson, NY, USA, 2000. [Google Scholar]
- Lynch, M. The Similarity Index and DNA Fingerprinting. Mol. Biol. Evol. 1990, 7, 478–484. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising How the Computer Program CERVUS Accommodates Genotyping Error Increases Success in Paternity Assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research—An Update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Liu, J.X. StructureSelector: A Web-Based Software to Select and Visualize the Optimal Number of Clusters Using Multiple Methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A Cluster Matching and Permutation Program for Dealing with Label Switching and Multimodality in Analysis of Population Structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. Pophelper: An R Package and Web App to Analyse and Visualize Population Structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Locus | N | Na | Ne | I | Ho | He | F | PIC | PD | Nra |
|---|---|---|---|---|---|---|---|---|---|---|
| BPPCT001 | 885 | 15 | 4.1 | 1.81 | 0.5 | 0.76 | 0.34 | 0.73 | 0.92 | 8 |
| BPPCT007 | 935 | 14 | 2.74 | 1.24 | 0.5 | 0.64 | 0.21 | 0.57 | 0.8 | 11 |
| BPPCT015 | 871 | 11 | 2.24 | 1.15 | 0.37 | 0.55 | 0.34 | 0.5 | 0.75 | 9 |
| BPPCT017 | 939 | 19 | 2.44 | 1.23 | 0.46 | 0.59 | 0.22 | 0.52 | 0.76 | 17 |
| BPPCT038 | 949 | 13 | 2.38 | 1.21 | 0.44 | 0.58 | 0.24 | 0.53 | 0.77 | 10 |
| CPDCT045 | 909 | 10 | 2.69 | 1.13 | 0.49 | 0.63 | 0.22 | 0.55 | 0.78 | 7 |
| CPPCT006 | 921 | 9 | 2.47 | 1.03 | 0.44 | 0.6 | 0.26 | 0.53 | 0.77 | 6 |
| EPPCU5176 | 948 | 12 | 2.84 | 1.24 | 0.5 | 0.65 | 0.23 | 0.59 | 0.82 | 9 |
| UDP-005 | 841 | 15 | 2.36 | 1.21 | 0.4 | 0.58 | 0.3 | 0.54 | 0.78 | 11 |
| UDP-022 | 894 | 8 | 3.01 | 1.32 | 0.42 | 0.67 | 0.38 | 0.63 | 0.85 | 4 |
| UDP-409 | 926 | 10 | 1.74 | 0.88 | 0.32 | 0.42 | 0.24 | 0.4 | 0.64 | 7 |
| UDP-412 | 932 | 12 | 3.35 | 1.45 | 0.53 | 0.7 | 0.24 | 0.66 | 0.87 | 8 |
| Sample/SSR | BPPCT001 | BPPCT007 | BPPCT015 | BPPCT017 | BPPCT038 | CPDCT045 | CPPCT006 | EPPCU5176 | UDP-005 | UDP-022 | UDP-409 | UDP-412 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AB/6 | 170, 190 | 169, 177 | - - | 157, 161 | 125, 129 | 178, 184 | 220, 224 | 205, 205 | 162, 174 | 167, 167 | 158, 168 | 154, 154 |
| Chui_Huang_Tao | 182, 182 | 177, 177 | 186, 186 | 151, 151 | 131, 149 | 176, 176 | - - | 193, 193 | 157, 157 | 165, 169 | 181, 181 | 152, 152 |
| Citation | 162, 166 | 171, 177 | 198, 198 | 161, 161 | 123, 131 | 176, 178 | 220, 226 | 185, 197 | 191, 191 | 165, 165 | 158, 158 | 152, 160 |
| Dourado | 186, 188 | 161, 177 | 180, 180 | 151, 165 | 123, 127 | 176, 178 | 208, 220 | 197, 207 | 172, 191 | 165, 167 | 158, 158 | 152, 158 |
| Ferganensis | 182, 182 | 183, 183 | - - | 161, 161 | 123, 123 | 178, 178 | 220, 220 | 209, 209 | 193, 195 | 165, 165 | 183, 183 | 162, 162 |
| Fiorenza | 188, 188 | 177, 183 | 198, 198 | 161, 185 | 123, 123 | 180, 180 | 220, 220 | 197, 205 | 191, 191 | 165, 171 | 152, 158 | 158, 160 |
| Glowin_Star | 170, 190 | 179, 179 | 198, 198 | 134, 169 | 123, 123 | 176, 186 | 220, 224 | 207, 207 | 174, 189 | 165, 165 | 160, 183 | 156, 156 |
| IF_817023 | 162, 162 | - - | 186, 186 | 155, 161 | 123, 123 | 167, 180 | 216, 228 | 191, 191 | 174, 174 | - - | 183, 183 | 128, 133 |
| P1/12 | 186, 186 | 177, 198 | 198, 198 | 161, 161 | 123, 123 | 176, 176 | 220, 220 | 197, 207 | 176, 176 | 157, 157 | 158, 158 | 156, 158 |
| P5/645 | 182, 188 | 161, 177 | 186, 186 | 177, 179 | 135, 135 | 176, 180 | 208, 208 | 203, 207 | 189, 191 | 165, 171 | 158, 158 | 158, 158 |
| Queen_Ruby | 176, 184 | 179, 183 | 198, 200 | 161, 161 | 123, 123 | 178, 180 | 208, 220 | 201, 211 | 189, 191 | 165, 171 | 158, 158 | 152, 156 |
| Red_Robin | 182, 182 | 165, 175 | 198, 198 | 149, 179 | 129, 135 | 176, 180 | 208, 208 | 197, 201 | 191, 191 | 165, 171 | 152, 158 | 152, 158 |
| Shan_Dong | 184, 184 | 161, 177 | 198, 198 | 149, 179 | 123, 141 | 176, 180 | - - | 197, 199 | 174, 191 | - - | 158, 181 | 152, 158 |
| XIAGUANG_a | 186, 186 | 167, 183 | 198, 198 | 159, 161 | 123, 123 | 176, 176 | 208, 208 | 201, 201 | 174, 176 | 171, 171 | 158, 160 | - - |
| Type of Pedigree | N° | N Match | % Match |
|---|---|---|---|
| P1 × P2 | 47 | 16 | 34.0 |
| P × Self | 14 | 5 | 35.7 |
| P op | 59 | 42 | 71.2 |
| Clones | 29 | 17 | 55.2 |
| N° Tot | 149 | 80 | 53.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vendramin, E.; da Silva Linge, C.; Bassi, D.; Micali, S.; Chietera, G.; Dettori, M.T.; Aramini, V.; Giovinazzi, J.; Pacheco, I.; Rossini, L.; et al. Genebank Management Through Microsatellite Markers: A Case Study in Two Italian Peach Germplasm Collections. Plants 2025, 14, 2139. https://doi.org/10.3390/plants14142139
Vendramin E, da Silva Linge C, Bassi D, Micali S, Chietera G, Dettori MT, Aramini V, Giovinazzi J, Pacheco I, Rossini L, et al. Genebank Management Through Microsatellite Markers: A Case Study in Two Italian Peach Germplasm Collections. Plants. 2025; 14(14):2139. https://doi.org/10.3390/plants14142139
Chicago/Turabian StyleVendramin, Elisa, Cássia da Silva Linge, Daniele Bassi, Sabrina Micali, Giorgiana Chietera, Maria Teresa Dettori, Valeria Aramini, Jessica Giovinazzi, Igor Pacheco, Laura Rossini, and et al. 2025. "Genebank Management Through Microsatellite Markers: A Case Study in Two Italian Peach Germplasm Collections" Plants 14, no. 14: 2139. https://doi.org/10.3390/plants14142139
APA StyleVendramin, E., da Silva Linge, C., Bassi, D., Micali, S., Chietera, G., Dettori, M. T., Aramini, V., Giovinazzi, J., Pacheco, I., Rossini, L., & Verde, I. (2025). Genebank Management Through Microsatellite Markers: A Case Study in Two Italian Peach Germplasm Collections. Plants, 14(14), 2139. https://doi.org/10.3390/plants14142139