

The Dynamics of Long Terminal Repeat Retrotransposon Proliferation and Decay Drive the Evolution of Genome Size Variation in Capsicum

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Transposable Element Annotation and Classification
2.2. Comparative Genomic Analysis
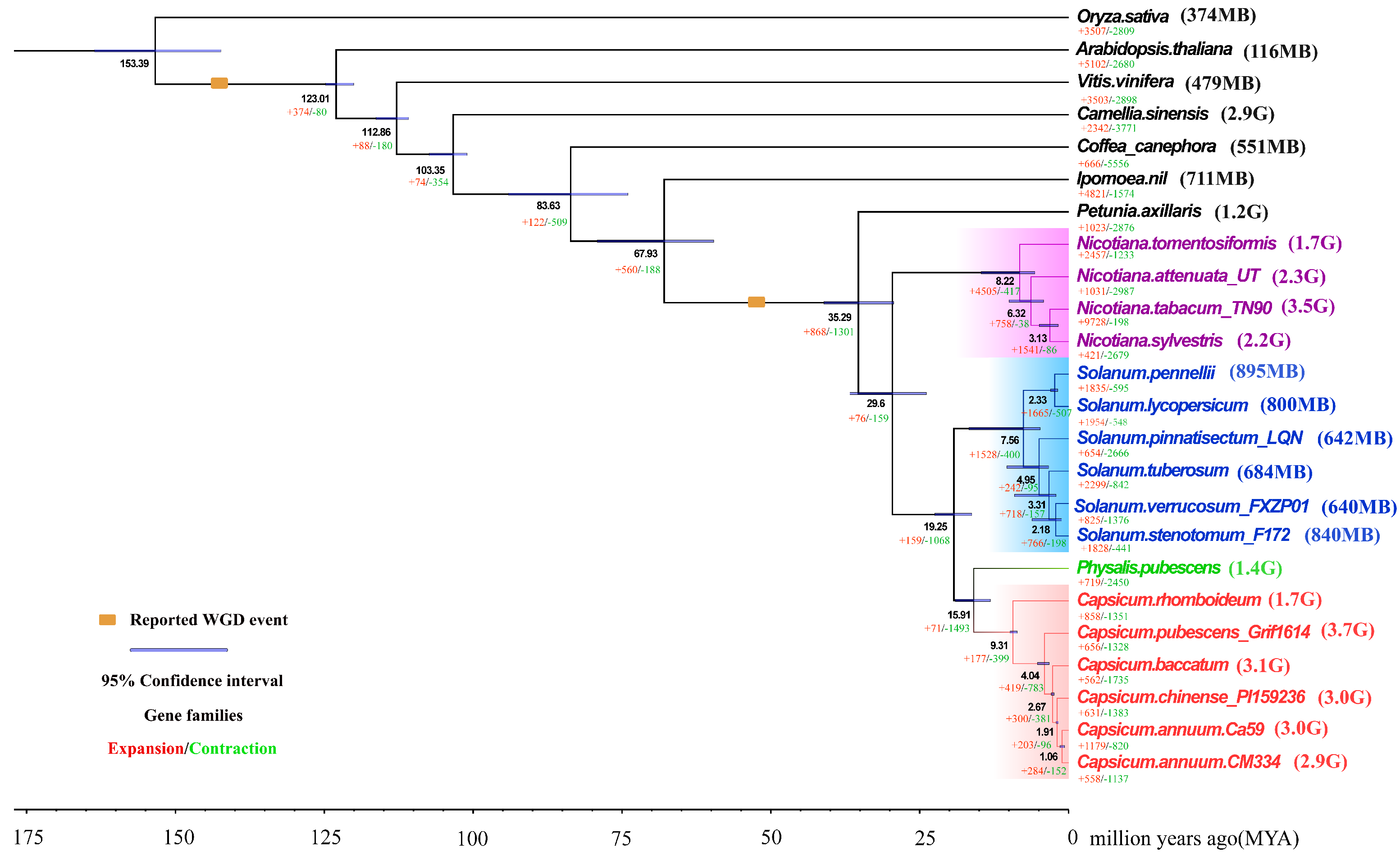
2.3. Phylogenetic Analysis
2.4. TE Dynamics Analysis
2.5. Transcriptional Activity Analysis
3. Results
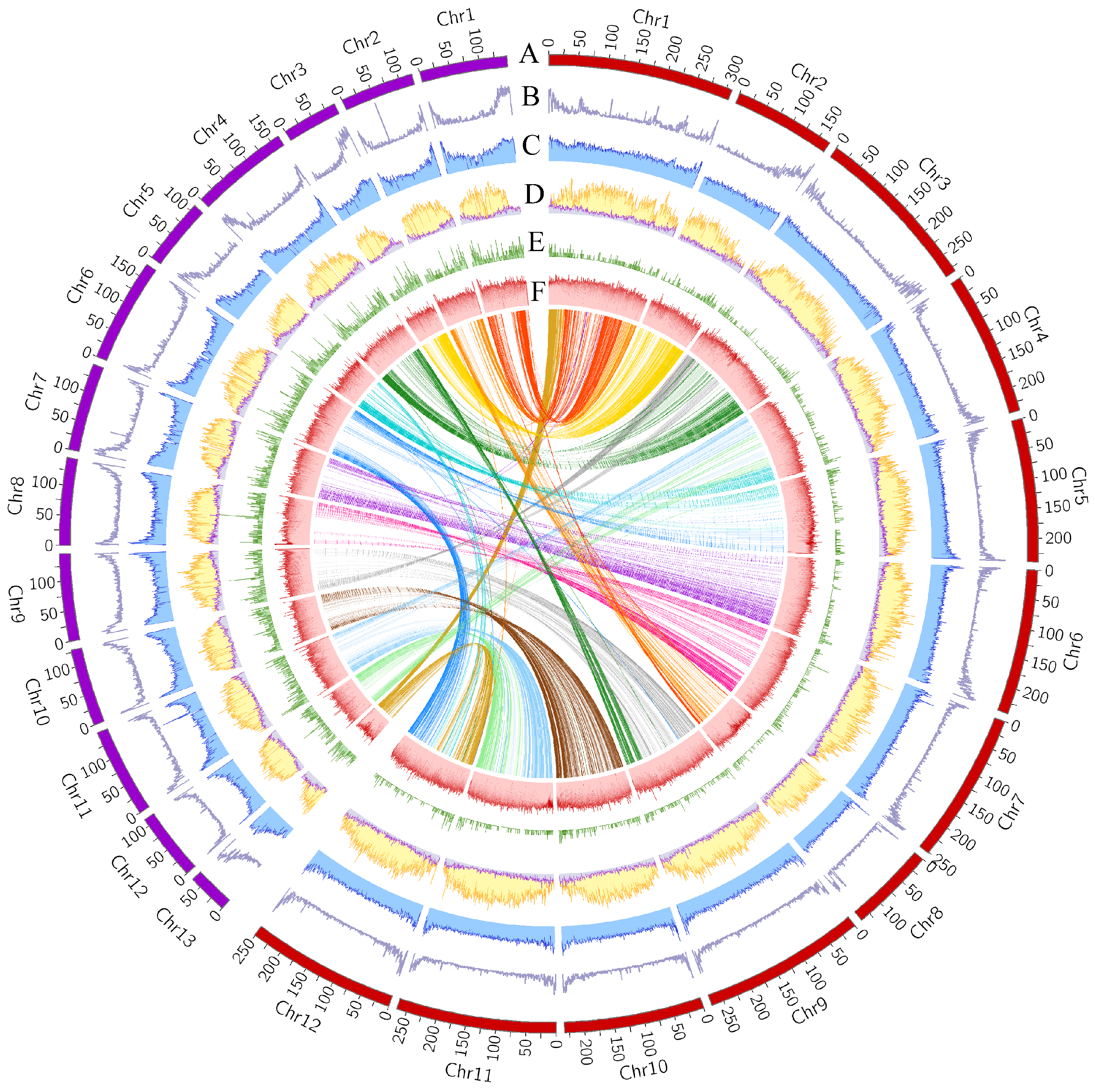
3.1. Comparative Genomics and Evolution of Capsicum Genomes with Divergent Sizes
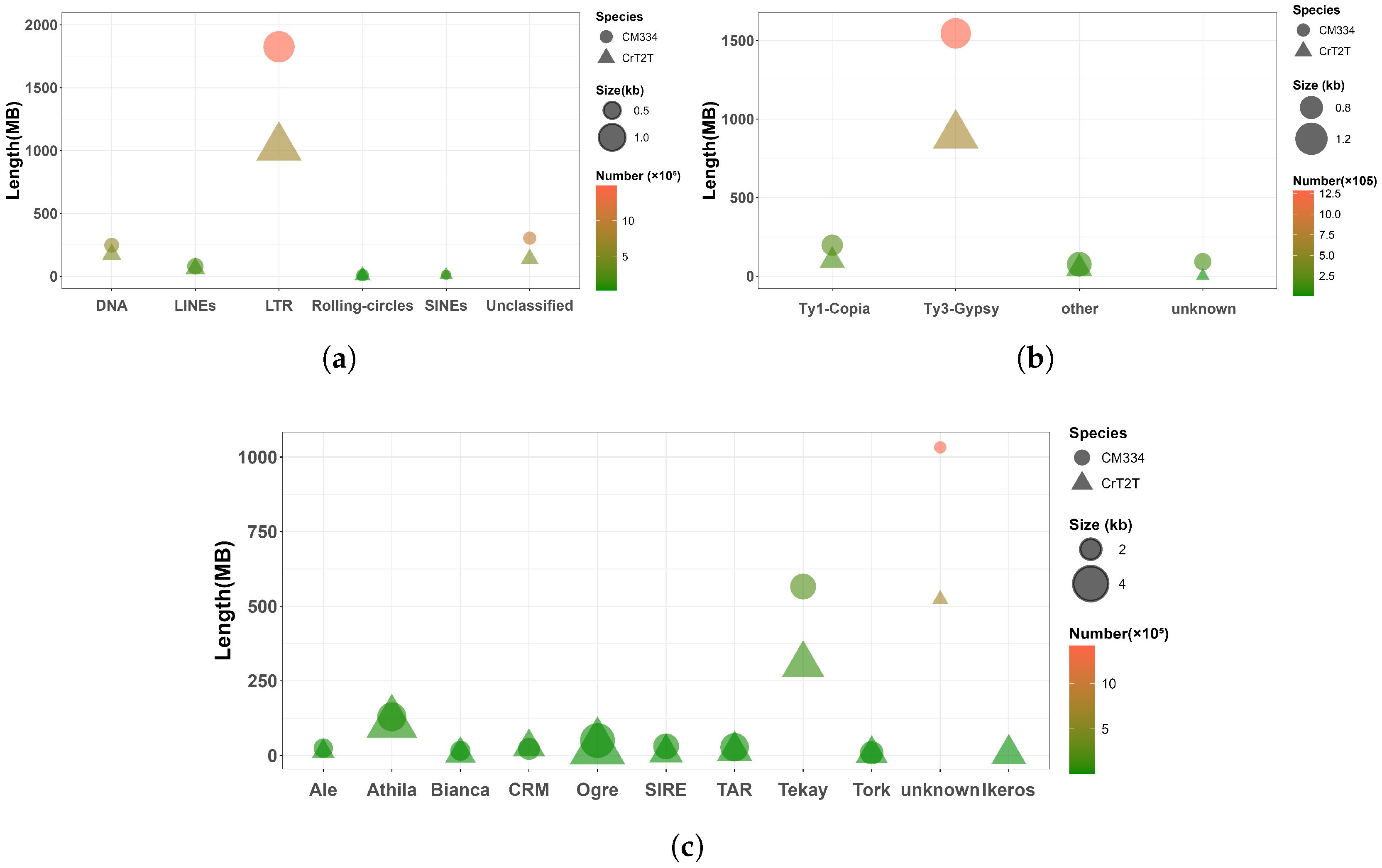
3.2. Transposable Element Composition and Abundance Correlate with Genome Size
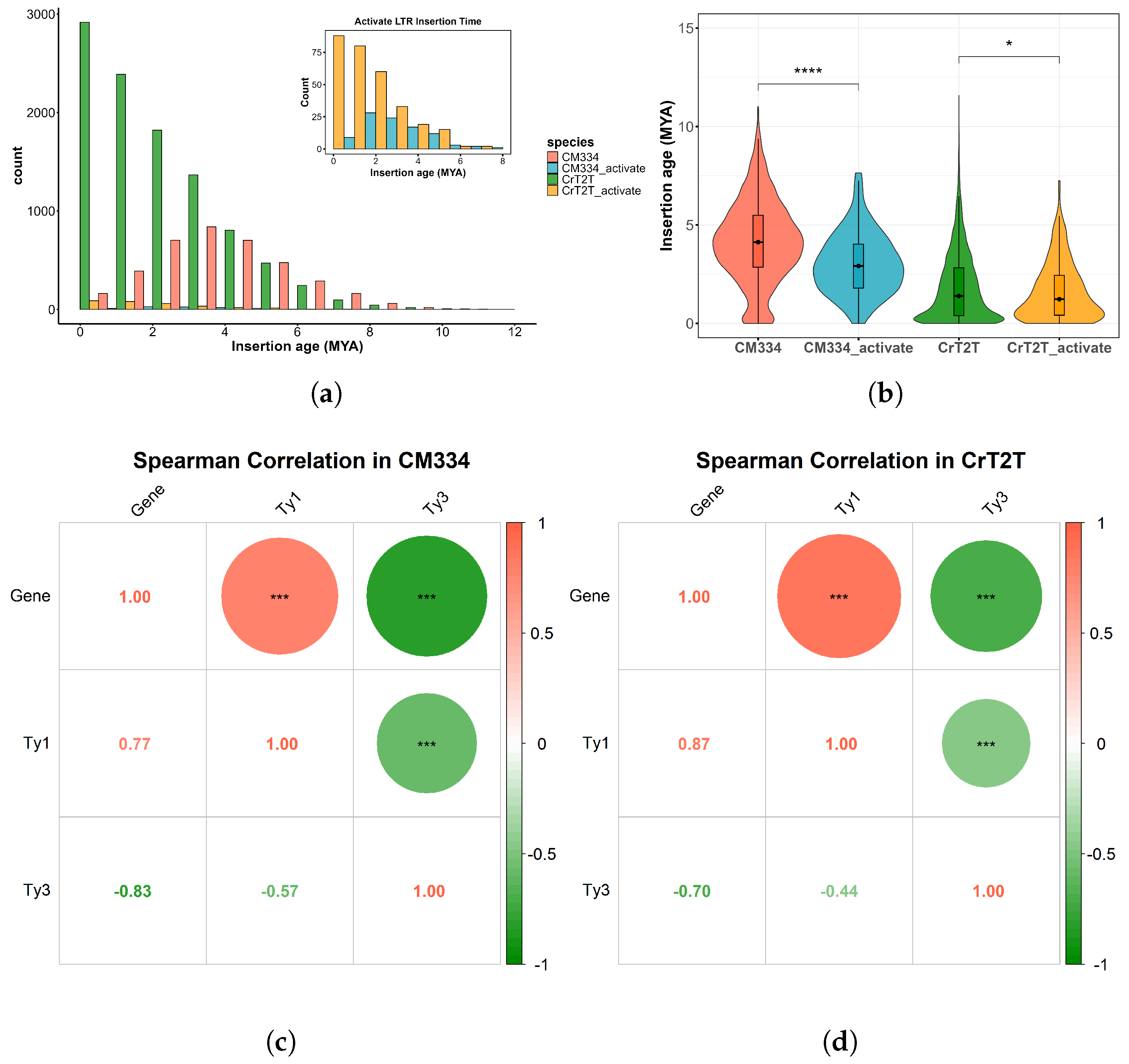
3.3. Transposable Element Insertion Patterns Reflect Dynamic Changes in Genome Size
3.4. Transposable Element Transcriptional Activity Identification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TE | Transposable element |
| LTR-RT | Long terminal repeat retrotransposon |
| LTR | Long terminal repeat |
| RT | reverse transcriptase |
| WGD | Whole genome duplication |
| CrT2T | Capsicum rhomboideum |
| CM334 | Capsicum annuum cv. CM334 |
References
- Swift, H. The constancy of desoxyribose nucleic acid in plant nuclei. Proc. Natl. Acad. Sci. USA 1950, 36, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Leitch, I. Plant genome size research: A field in focus. Ann. Bot. 2005, 95, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Leitch, I.J. The Plant DNA C-values database (release 7.1). New Phytol. 2020, 226, 301–305. [Google Scholar] [CrossRef]
- Grover, C.E.; Wendel, J.F. Recent insights into mechanisms of genome size change in plants. J. Bot. 2010, 2010, 382732. [Google Scholar] [CrossRef]
- Gregory, T.R. Synergy between sequence and size in large-scale genomics. Nat. Rev. Genet. 2005, 6, 699–708. [Google Scholar] [CrossRef]
- Pellicer, J.; Hidalgo, O.; Dodsworth, S.; Leitch, I.J. Genome size diversity and its impact on the evolution of land plants. Genes 2018, 9, 88. [Google Scholar] [CrossRef]
- de Assis, R.; Baba, V.Y.; Cintra, L.A.; Gonçalves, L.S.A.; Rodrigues, R.; Vanzela, A.L.L. Genome relationships and LTR-retrotransposon diversity in three cultivated Capsicum L. (Solanaceae) species. BMC Genom. 2020, 21, 237. [Google Scholar] [CrossRef]
- Heslop-Harrison, J.; Schwarzacher, T. Organisation of the plant genome in chromosomes. Plant J. 2011, 66, 18–33. [Google Scholar] [CrossRef]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef]
- Meng, H.; Feng, J.; Bai, T.; Jian, Z.; Chen, Y.; Wu, G. Genome-wide analysis of short interspersed nuclear elements provides insight into gene and genome evolution in citrus. DNA Res. 2020, 27, dsaa004. [Google Scholar] [CrossRef]
- Kang, H.; Zhu, D.; Lin, R.; Opiyo, S.O.; Jiang, N.; Shiu, S.H.; Wang, G.L. A novel method for identifying polymorphic transposable elements via scanning of high-throughput short reads. DNA Res. 2016, 23, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Wang, P.; Liu, F.; Liu, Z.; Zhao, G.; Xu, Z.; Pei, L.; Grover, C.E.; Wendel, J.F.; et al. Comparative genome analyses highlight transposon-mediated genome expansion and the evolutionary architecture of 3D genomic folding in cotton. Mol. Biol. Evol. 2021, 38, 3621–3636. [Google Scholar] [CrossRef] [PubMed]
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Albert, V.A.; Leebens-Mack, J.; Bell, C.D.; Paterson, A.H.; Zheng, C.; Sankoff, D.; de Pamphilis, C.W.; Wall, P.K.; Soltis, P.S. Polyploidy and angiosperm diversification. Am. J. Bot. 2009, 96, 336–348. [Google Scholar] [CrossRef]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and evolution of a minute plant genome. Nature 2013, 498, 94–98. [Google Scholar] [CrossRef]
- Kellogg, E.A.; Bennetzen, J.L. The evolution of nuclear genome structure in seed plants. Am. J. Bot. 2004, 91, 1709–1725. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Sanmiguel, P.; Bennetzen, J.L. Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Ann. Bot. 1998, 82, 37–44. [Google Scholar] [CrossRef]
- Vitte, C.; Panaud, O. LTR retrotransposons and flowering plant genome size: Emergence of the increase/decrease model. Cytogenet. Genome Res. 2005, 110, 91–107. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 2004, 5, R79. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, K.; Srinivasan, S.; Bureau, T. Survey of transposable elements from rice genomic sequences. Plant J. 2001, 25, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Devos, K.M.; Bennetzen, J.L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14, 860–869. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Wang, J.; Gong, Z.; Han, G.Z. Plants acquired a major retrotransposon horizontally from fungi during the conquest of land. New Phytol. 2021, 232, 11–16. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Yeom, S.I.; Kim, Y.M.; Lee, J.M.; Lee, H.A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.T.; et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270–278. [Google Scholar] [CrossRef]
- Qin, C.; Yu, C.; Shen, Y.; Fang, X.; Chen, L.; Min, J.; Cheng, J.; Zhao, S.; Xu, M.; Luo, Y.; et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. USA 2014, 111, 5135–5140. [Google Scholar] [CrossRef]
- Chen, W.; Wang, X.; Sun, J.; Wang, X.; Zhu, Z.; Ayhan, D.H.; Yi, S.; Yan, M.; Zhang, L.; Meng, T.; et al. Two telomere-to-telomere gapless genomes reveal insights into Capsicum evolution and capsaicinoid biosynthesis. Nat. Commun. 2024, 15, 4295. [Google Scholar] [CrossRef]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 5, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018, 176, 1410–1422. [Google Scholar] [CrossRef]
- Zhang, R.G.; Li, G.Y.; Wang, X.L.; Dainat, J.; Wang, Z.X.; Ou, S.; Ma, Y. TEsorter: An accurate and fast method to classify LTR-retrotransposons in plant genomes. Hortic. Res. 2022, 9, uhac017. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Zeng, X.; Xu, Z.; Taranto, A.; Lomas, J.S.; Zhang, Y.; Huang, Y.; Wang, Y.; Yim, W.C.; et al. JCVI: A versatile toolkit for comparative genomics analysis. Imeta 2024, 3, e211. [Google Scholar] [CrossRef]
- Sun, P.; Jiao, B.; Yang, Y.; Shan, L.; Li, T.; Li, X.; Xi, Z.; Wang, X.; Liu, J. WGDI: A user-friendly toolkit for evolutionary analyses of whole-genome duplications and ancestral karyotypes. Mol. Plant 2022, 15, 1841–1851. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 May 2025).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef]
- Carrizo García, C.; Barfuss, M.H.; Sehr, E.M.; Barboza, G.E.; Samuel, R.; Moscone, E.A.; Ehrendorfer, F. Phylogenetic relationships, diversification and expansion of chili peppers (Capsicum, Solanaceae). Ann. Bot. 2016, 118, 35–51. [Google Scholar] [CrossRef]
- Zhang, K.; Yu, H.; Zhang, L.; Cao, Y.; Li, X.; Mei, Y.; Wang, X.; Zhang, Z.; Li, T.; Jin, Y.; et al. Transposon proliferation drives genome architecture and regulatory evolution in wild and domesticated peppers. Nat. Plants 2025, 11, 359–375. [Google Scholar] [CrossRef]
- Bliss, B.J.; Suzuki, J.Y. Genome size in Anthurium evaluated in the context of karyotypes and phenotypes. AoB Plants 2012, 2012, pls006. [Google Scholar] [CrossRef]
- Deng, S. The origin of genetic and metabolic systems: Evolutionary structuralinsights. Heliyon 2023, 9, e14466. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.S.; Grover, C.E.; Wendel, J.F. Repeated big bangs and the expanding universe: Directionality in plant genome size evolution. Plant Sci. 2008, 174, 557–562. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, S.; Bailey, J.A.; Sahinalp, S.C.; Alkan, C.; Tuzun, E.; Green, E.D.; Eichler, E.E.; Program, N.C.S. Analysis of primate genomic variation reveals a repeat-driven expansion of the human genome. Genome Res. 2003, 13, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Cronn, R.C.; Spencer Johnston, J.; James Price, H. Feast and famine in plant genomes. Genetica 2002, 115, 37–47. [Google Scholar] [CrossRef]
- Zhang, Q.J.; Gao, L.Z. Rapid and recent evolution of LTR retrotransposons drives rice genome evolution during the speciation of AA-genome Oryza species. G3 Genes Genomes Genet. 2017, 7, 1875–1885. [Google Scholar] [CrossRef]
- Mascagni, F.; Giordani, T.; Ceccarelli, M.; Cavallini, A.; Natali, L. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genom. 2017, 18, 634. [Google Scholar] [CrossRef]
- Benoit, M.; Jenike, K.M.; Satterlee, J.W.; Ramakrishnan, S.; Gentile, I.; Hendelman, A.; Passalacqua, M.J.; Suresh, H.; Shohat, H.; Robitaille, G.M.; et al. Solanum pan-genetics reveals paralogues as contingencies in crop engineering. Nature 2025, 640, 135–145. [Google Scholar] [CrossRef]
- Guo, L.; Wang, X.; Ayhan, D.H.; Rhaman, M.S.; Yan, M.; Jiang, J.; Wang, D.; Zheng, W.; Mei, J.; Ji, W.; et al. Super pangenome of Vitis empowers identification of downy mildew resistance genes for grapevine improvement. Nature Genetics 2025, 57, 741–753. [Google Scholar] [CrossRef]
- Zhang, S.J.; Liu, L.; Yang, R.; Wang, X. Genome size evolution mediated by Gypsy retrotransposons in Brassicaceae. Genom. Proteom. Bioinform. 2020, 18, 321–332. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Size (Gb) | Chromosome Number | Gene Number | Total Gene Length (bp) | |
|---|---|---|---|---|
| Capsicum rhomboideum | 1.7 | 13 | 33,512 | 181,798,887 |
| Capsicum annuum cv. CM334 | 2.9 | 12 | 31,600 | 142,495,079 |
| Transcriptionally Active TEs | Transcriptionally Active Intact LTR-RTs | ||||||
|---|---|---|---|---|---|---|---|
| Genome | Ty1 | Ty3 | Pararetrovirus | LINE | Ty1 | Ty3 | |
| CrT2T | 981 | 730 | 89 | 0 | 177 | 121 | |
| CM334 | 687 | 358 | 44 | 182 | 73 | 24 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Liu, P.; Wang, S.; Yang, J.; Dai, L.; Zheng, J.; Wang, Y. The Dynamics of Long Terminal Repeat Retrotransposon Proliferation and Decay Drive the Evolution of Genome Size Variation in Capsicum. Plants 2025, 14, 2136. https://doi.org/10.3390/plants14142136
Liu Q, Liu P, Wang S, Yang J, Dai L, Zheng J, Wang Y. The Dynamics of Long Terminal Repeat Retrotransposon Proliferation and Decay Drive the Evolution of Genome Size Variation in Capsicum. Plants. 2025; 14(14):2136. https://doi.org/10.3390/plants14142136
Chicago/Turabian StyleLiu, Qian, Pinbo Liu, Shenghui Wang, Jian Yang, Liangying Dai, Jingyuan Zheng, and Yunsheng Wang. 2025. "The Dynamics of Long Terminal Repeat Retrotransposon Proliferation and Decay Drive the Evolution of Genome Size Variation in Capsicum" Plants 14, no. 14: 2136. https://doi.org/10.3390/plants14142136
APA StyleLiu, Q., Liu, P., Wang, S., Yang, J., Dai, L., Zheng, J., & Wang, Y. (2025). The Dynamics of Long Terminal Repeat Retrotransposon Proliferation and Decay Drive the Evolution of Genome Size Variation in Capsicum. Plants, 14(14), 2136. https://doi.org/10.3390/plants14142136