
Phylogenomic Analyses Reveal Species Relationships and Phylogenetic Incongruence with New Member Detected in Allium Subgenus Cyathophora


Abstract
1. Introduction
2. Results
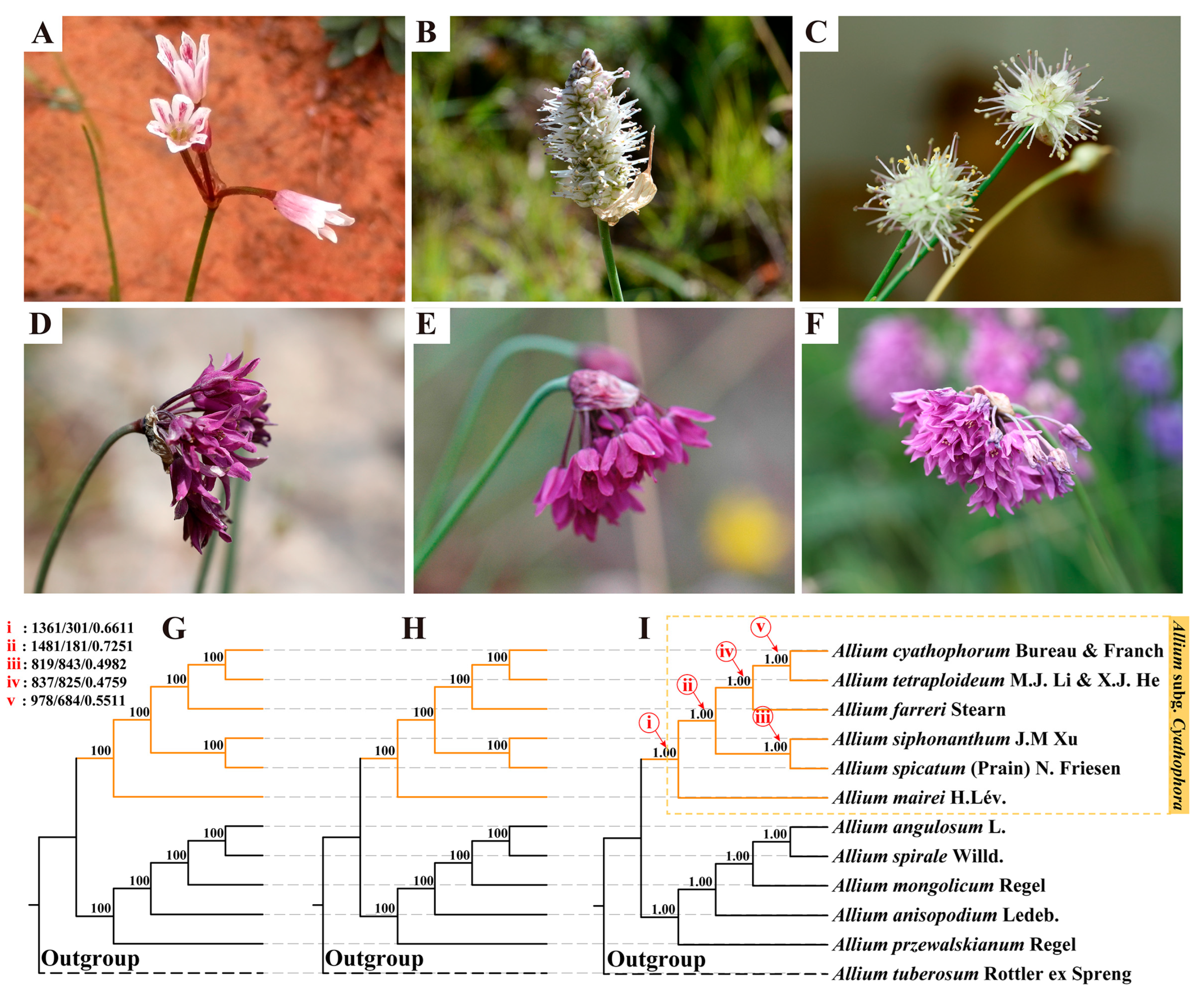
2.1. Summary of Morphological Characteristics
2.2. Transcriptome Assembly and Phylogeny Results
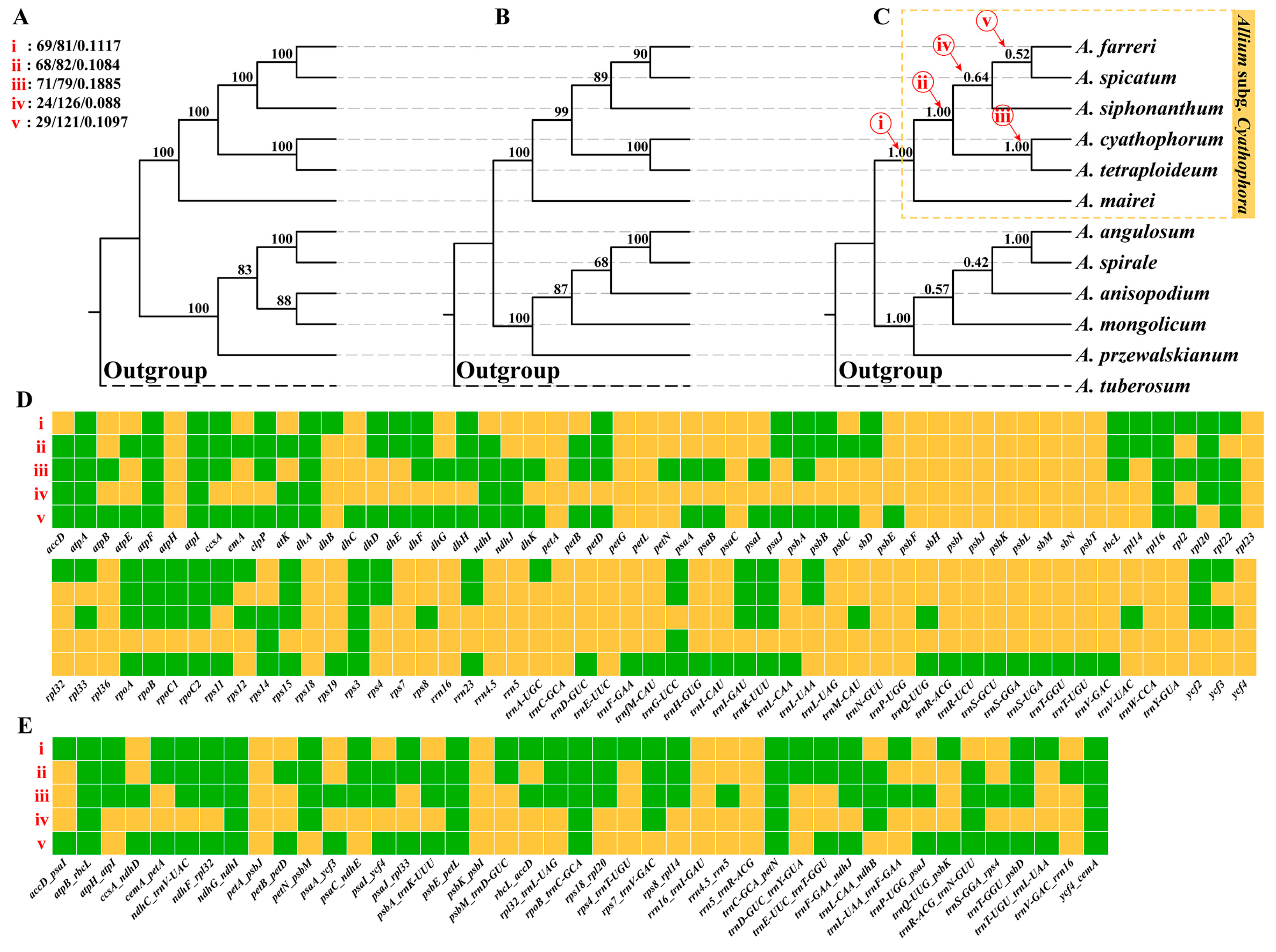
2.3. Phylogenetic Results Based on Plastome Data
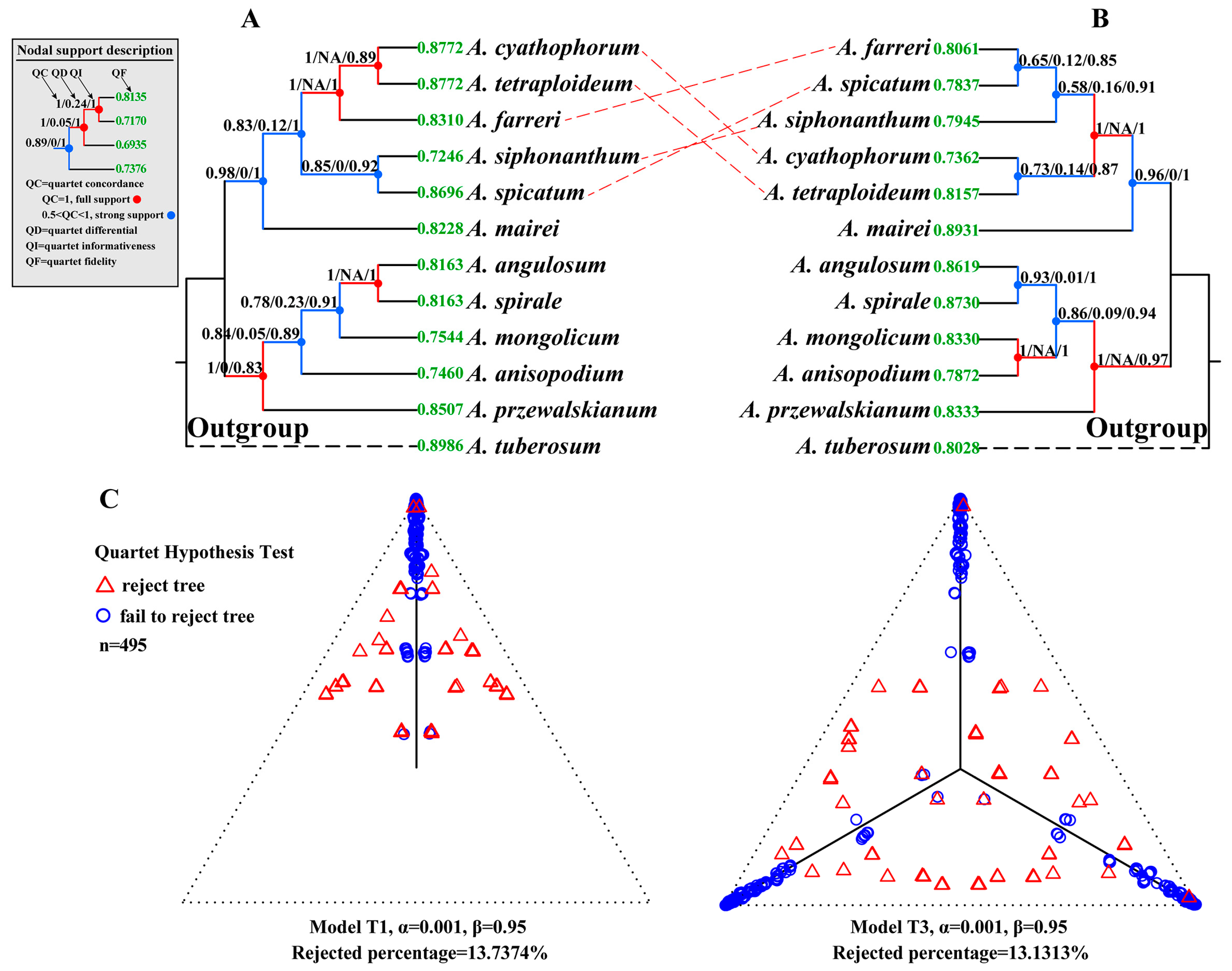
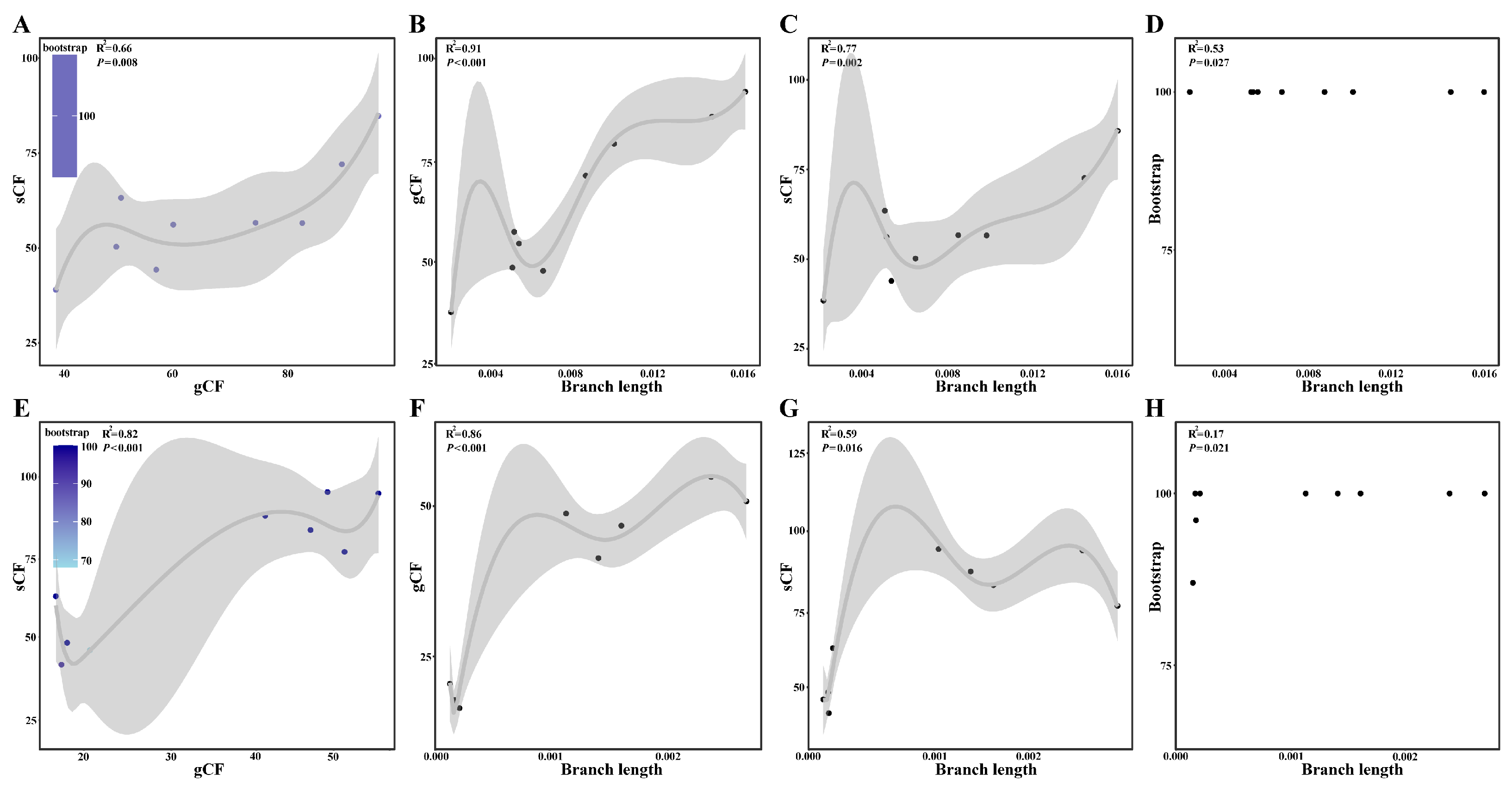
2.4. Concordance Analysis
2.5. Detection of ILS
2.6. Reticulation Identification
3. Discussion
3.1. Phylogenomic Data Revealed New Species Relationships in Subg. Cyathophora
3.2. Underlying Causes for Phylogenetic Discordances in Subg. Cyathophora
4. Materials and Methods
4.1. Sampling, Morphological Observation, and Transcriptome Sequencing
4.2. Transcriptome Assembly and Low-Copy Gene Identification
4.3. Phylogenetic Reconstruction and ICA Score Calculation
4.4. Whole-Genome Resequencing, Plastome Assembly, and Phylogenetic Analyses
4.5. Phylogenetic Discordance Analysis
4.6. Estimation of Incomplete Lineage Sorting
4.7. Reticulate Evolution Detection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chase, M.W.; Reveal, J.L. A phylogenetic classification of the land plants to accompany APG III. Bot. J. Linn. Soc. 2009, 161, 122–127. [Google Scholar] [CrossRef]
- Herden, T.; Hanelt, P.; Friesen, N. Phylogeny of Allium L. subgenus Anguinum (G. Don. ex W.D.J. Koch) N. Friesen (Amaryllidaceae). Mol. Phylogenet. Evol. 2016, 95, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, R.; Kington, S.; Friesen, N.; Fritsch, R.M.; Snijman, D.A.; Marcucci, R.; Silverstone-Sopkin, P.A.; Brullo, S. World Checklist of Amaryllidaceae. Facilitated by the Roy. Bot. Gard., Kew. 2021. Available online: http://apps.kew.org/wcsp/ (accessed on 27 February 2025).
- Block, E. Garlic and Other Alliums: The Lore and the Science; Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Fritsch, R.M.; Friesen, N. Evolution, domestication and taxonomy. In Allium Crop Science: Recent Advances; Rabinowitch, H.D., Currah, L., Eds.; CABI Publishing: Wallingford, UK, 2002; pp. 5–30. [Google Scholar]
- Friesen, N.; Fritsch, R.M.; Blattner, F.R. Phylogeny and intrageneric classification of Allium (Alliaceae) based on nuclear ribosomal DNA ITS sequences. Aliso 2006, 22, 372–395. [Google Scholar] [CrossRef]
- Jang, J.E.; Baasanmunkh, S.; Nyamgerel, N.; Oh, S.Y.; Song, J.H.; Yusupov, Z.; Tojibaev, K.; Choi, H.J. Flower morphology of Allium (Amaryllidaceae) and its systematic significance. Plant Divers. 2023, 46, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Oh, B.U. A partial revision of Allium (Amaryllidaceae) in Korea and north-eastern China. Bot. J. Linn. Soc. 2011, 167, 153–211. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Driscoll, H.E.; Specht, C.D. A molecular phylogeny of the wild onions (Allium: Alliaceae) with a focus on the western North American center of diversity. Mol. Phylogenet. Evol. 2008, 47, 1157–1172. [Google Scholar] [CrossRef]
- Li, M.J.; Tan, J.B.; Xie, D.F.; Huang, D.Q.; Gao, Y.D.; He, X.J. Revisiting the evolutionary events in Allium subgenus Cyathophora (Amaryllidaceae): Insights into the effect of the Hengduan Mountains Region (HMR) uplift and quaternary climatic fluctuations to the environmental changes in the Qinghai-Tibet Plateau. Mol. Phylogenet. Evol. 2016, 94, 802–813. [Google Scholar] [CrossRef]
- Li, M.J.; Zheng, Z.Y.; Liu, J.C.; Yang, Y.Z.; Ren, G.P.; Ru, D.F.; Zhang, S.Z.; Du, X.; Ma, T.; Milne, R.; et al. Evolutionary origin of a tetraploid Allium species on the Qinghai-Tibet Plateau. Mol. Ecol. 2021, 30, 5780–5795. [Google Scholar] [CrossRef]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium species in section Daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef]
- Xie, D.F.; Tan, J.B.; Yu, Y.; Gui, L.J.; Su, D.M.; Zhou, S.D.; He, X.J. Insights into phylogeny, age and evolution of Allium (Amaryllidaceae) based on the whole plastome sequences. Ann. Bot. 2020, 125, 1039–1055. [Google Scholar] [CrossRef]
- Fu, X.; Xie, D.F.; Zhou, Y.Y.; Cheng, R.Y.; Zhang, X.Y.; Zhou, S.D.; He, X.J. Phylogeny and adaptive evolution of subgenus Rhizirideum (Amaryllidaceae, Allium) based on plastid genomes. BMC Plant Biol. 2023, 23, 70. [Google Scholar] [CrossRef] [PubMed]
- Friesen, N.; Herden, T.; Leweke, M.; Grützmacher, L.; Fragman-Sapir, O.; Hurka, H.; Blattner, F.; Fritsch, R.M. Dated phylogeny, phylogeography, and classification of Allium subgenus Amerallium (Amaryllidaceae) from the old world, based on six DNA fragments. Taxon 2024, 73, 971–991. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, G.; Li, M. Phylotranscriptomic discordance is best explained by incomplete lineage sorting within Allium subgenus Cyathophora and thus hemiplasy accounts for interspecific trait transition. Plant Divers. 2024, 46, 28–38. [Google Scholar] [CrossRef]
- Li, M.J.; Liu, J.Q.; Guo, X.L.; Xiao, Q.Y.; He, X.J. Taxonomic revision of Allium Cyathophorum (Amaryllidaceae). Phytotaxa 2019, 415, 240–246. [Google Scholar] [CrossRef]
- Friesen, N.; Fritsch, R.M.; Pollner, S.; Blattner, F.R. Molecular and morphological evidence for an origin of the aberrant genus Milula within Himalayan species of Allium (Alliaceae). Mol. Phylogenet. Evol. 2000, 17, 209–218. [Google Scholar] [CrossRef]
- Tang, H.G.; Meng, L.H.; Ao, S.Q.; Liu, J.Q. Origin of the Qinghai-Tibetan Plateau endemic Milula (Liliaceae): Further insights from karyological comparisons with Allium. Caryologia 2005, 58, 320–331. [Google Scholar] [CrossRef]
- Xu, J.M. Allium siphonanthum J.M.Xu. Fl. Reipubl. Popularis Sin. 1980, 14, 284. [Google Scholar]
- Li, Q.Q. Molecular Systematics of the Genus Allium L. (Amaryllidaceae: Allieae). Doctoral Thesis, Sichuan University, Chengdu, China, 2010. [Google Scholar]
- Meleshko, O.; Martin, M.D.; Korneliussen, T.S.; Schröck, C.; Lamkowski, P.; Schmutz, J.; Healey, A.; Piatkowski, B.T.; Shaw, A.J.; Weston, D.J.; et al. Extensive genome-wide phylogenetic discordance is due to incomplete lineage sorting and not ongoing introgression in a rapidly radiated bryophyte genus. Mol. Biol. Evol. 2021, 38, 2750–2766. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, X.; Herre, E.A.; McKey, D.; Machado, C.A.; Yu, W.B.; Cannon, C.H.; Arnold, M.L.; Pereira, R.A.S.; Ming, R.; et al. Genomic evidence of prevalent hybridization throughout the evolutionary history of the fig-wasp pollination mutualism. Nat. Commun. 2021, 12, 718. [Google Scholar] [CrossRef]
- Cheng, L.; Han, Q.; Chen, F.; Balbuena, T.S.; Zhao, Y. Phylogenomics as an effective approach to untangle cross-species hybridization events: A case study in the family Nymphaeaceae. Front. Genet. 2022, 13, 1031705. [Google Scholar] [CrossRef]
- Feng, Y.; Xiang, X.; Akhter, D.; Pan, R.; Fu, Z.; Jin, X. Mitochondrial phylogenomics of Fagales provides insights into plant mitogenome mosaic evolution. Front. Plant Sci. 2021, 12, 762195. [Google Scholar] [CrossRef]
- Xia, X.M.; Yang, M.Q.; Li, C.L.; Huang, S.X.; Jin, W.T.; Shen, T.T.; Wang, F.; Li, X.H.; Yoichi, W.; Zhang, L.H.; et al. Spatiotemporal evolution of the global species diversity of Rhododendron. Mol. Biol. Evol. 2022, 39, msab314. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.F.; Li, J.; Sun, J.H.; Cheng, R.Y.; Wang, Y.; Song, B.N.; He, X.J.; Zhou, S.D. Peering through the hedge: Multiple datasets yield insights into the phylogenetic relationships and incongruences in the tribe Lilieae (Liliaceae). Mol. Phylogenet. Evol. 2024, 200, 108182. [Google Scholar] [CrossRef]
- Xu, L.; Song, Z.; Li, T.; Jin, Z.; Zhang, B.; Du, S.; Liao, S.; Zhong, X.; Chen, Y. New insights into the phylogeny and infrageneric taxonomy of Saussurea based on hybrid capture phylogenomics (Hyb-Seq). Plant Divers. 2025, 47, 21–33. [Google Scholar] [CrossRef] [PubMed]
- He, X.J. Integrating high-volume molecular and morphological data into the evolutionary studies of Allium. Plant Divers. 2024, 46, 1–2. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, G.; Li, M. Incomplete lineage sorting and gene flow within Allium (Amaryllidaceae). Mol. Phylogenet. Evol. 2024, 195, 108054. [Google Scholar] [CrossRef]
- Huang, D.Q. Molecular Systematics of Allium subgenus Cyathophora, Sections Bromatorrhiza and Sikkimensia (Amaryllidaceae) and Intraspecific Genetic Differentiation of A. wallichii. Doctoral Thesis, Sichuan University, Chengdu, China, 2014. [Google Scholar]
- Yang, L.H.; Harris, A.J.; Wen, F.; Li, Z.; Feng, C.; Kong, H.; Kang, M. Phylogenomic analyses reveal an allopolyploid origin of Core Didymocarpinae (Gesneriaceae) followed by rapid radiation. Syst. Biol. 2023, 72, 1064–1083. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.G.; Ren, Y.B.; Sun, H. Introgression and incomplete lineage sorting blurred phylogenetic relationships across the genomes of sclerophyllous oaks from southwest China. Cladistics 2024, 40, 357–373. [Google Scholar] [CrossRef]
- Naranjo, A.A.; Edwards, C.E.; Gitzendanner, M.A.; Soltis, D.E.; Soltis, P.S. Abundant incongruence in a clade endemic to a biodiversity hotspot: Phylogenetics of the scrub mint clade (Lamiaceae). Mol. Phylogenet. Evol. 2024, 192, 108014. [Google Scholar] [CrossRef]
- Taylor, S.A.; Larson, E.L. Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat. Ecol. Evol. 2019, 3, 170–177. [Google Scholar] [CrossRef]
- Rose, J.P.; Toledo, C.A.P.; Lemmon, E.M.; Lemmon, A.R.; Sytsma, K.J. Out of sight, out of mind: Widespread nuclear and plastid-nuclear discordance in the flowering plant genus Polemonium (Polemoniaceae) suggests widespread historical gene flow despite limited nuclear signal. Syst. Biol. 2021, 70, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Talavera, A.; Nie, Z.L.; Ma, Z.Y.; Ma, Z.-Y.; Johnson, G.; Ickert-Bond, S.M.; Zimmer, E.A.; Wen, J. Phylogenomic analyses using a new 1013-gene Vitaceae bait-set support major groups of North American Vitis. Mol. Phylogenet. Evol. 2023, 186, 107866. [Google Scholar] [CrossRef] [PubMed]
- Edelman, N.B.; Frandsen, P.B.; Miyagi, M.; Clavijo, B.; Davey, J.; Dikow, R.B.; García-Accinelli, G.; Van Belleghem, S.M.; Patterson, N.; Neafsey, D.E.; et al. Genomic architecture and introgression shape a butterfly radiation. Science 2019, 366, 594–599. [Google Scholar] [CrossRef]
- Dong, W.; Li, E.; Liu, Y.; Xu, C.; Wang, Y.; Liu, K.; Cui, X.; Sun, J.; Suo, Z.; Zhang, Z.; et al. Phylogenomic approaches untangle early divergences and complex diversifications of the olive plant family. BMC Biol. 2022, 20, 92. [Google Scholar] [CrossRef]
- Cai, L.; Xi, Z.; Lemmon, E.M.; Lemmon, A.R.; Mast, A.; Buddenhagen, C.E.; Liu, L.; Davis, C.C. The Perfect Storm: Gene Tree Estimation Error, Incomplete Lineage Sorting, and Ancient Gene Flow Explain the Most Recalcitrant Ancient Angiosperm Clade, Malpighiales. Syst. Biol. 2021, 70, 491–507. [Google Scholar] [CrossRef]
- Whitney, K.D.; Ahern, J.R.; Campbell, L.G.; Albert, L.P.; King, M.S. Patterns of hybridization in plants. Perspect. Plant Ecol. Evol. Syst. 2010, 12, 175–182. [Google Scholar] [CrossRef]
- Liu, J.; Nie, Z.L.; Ren, C.; Su, C.; Wen, J. Phylogenomics of Aralia sect. Aralia (Araliaceae): Signals of hybridization and insights into its species delimitations and intercontinental biogeography. Mol. Phylogenet. Evol. 2023, 181, 107727. [Google Scholar]
- Marianne, B.; Erin, K.M.; Colin, N.D.; Claudia, S.L. Detectability of varied hybridization scenarios using genome-scale hybrid detection methods. arXiv 2023. [Google Scholar] [CrossRef]
- Ma, Y.; Mao, X.; Wang, J.; Zhang, L.; Jiang, Y.; Geng, Y.; Ma, T.; Cai, L.; Huang, S.; Hollingsworth, P.; et al. Pervasive hybridization during evolutionary radiation of Rhododendron subgenus Hymenanthes in mountains of southwest China. Nat. Sci. Rev. 2022, 9, nwac276. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, M.H.; Li, J.L.; Zhang, Z.Y.; Wang, Y.F.; Chen, C.L.; Yang, Y.Z.; Liu, J.Q. Genomic evidence for homoploid hybrid speciation between ancestors of two different genera. Nat. Commun. 2022, 13, 1987. [Google Scholar] [CrossRef]
- Cardoni, S.; Piredda, R.; Denk, T.; Grimm, G.W.; Papageorgiou, A.C.; Schulze, E.D.; Scoppola, A.; Salehi Shanjani, P.; Suyama, Y.; Tomaru, N.; et al. 5S-IGS rDNA in wind-pollinated trees (Fagus L.) encapsulates 55 million years of reticulate evolution and hybrid origins of modern species. Plant J. 2022, 109, 909–926. [Google Scholar] [CrossRef] [PubMed]
- McLay, T.G.B.; Fowler, R.M.; Fahey, P.S.; Murphy, D.J.; Udovicic, F.; Cantrill, D.J.; Bayly, M.J. Phylogenomics reveals extreme gene tree discordance in a lineage of dominant trees: Hybridization, introgression, and incomplete lineage sorting blur deep evolutionary relationships despite clear species groupings in Eucalyptus subgenus Eudesmia. Mol. Phylogenet. Evol. 2023, 187, 107869. [Google Scholar] [CrossRef]
- Liu, J.Q.; Duan, Y.W.; Hao, G.; Ge, X.J.; Sun, H. Evolutionary history and underlying adaptation of alpine plants on the Qinghai-Tibet Plateau. J. Syst. Evol. 2014, 52, 241–249. [Google Scholar] [CrossRef]
- Marchese, C. Biodiversity hotspots: A shortcut for a more complicated concept. Glob. Ecol. Conserv. 2015, 3, 297–309. [Google Scholar] [CrossRef]
- Mosbrugger, V.; Favre, A.; Muellner-Riehl, A.N. Cenozoic evolution of geo-biodiversity in the Tibeto-Himalayan region. In Mountains, Climate, and Biodiversity; Hoorn, C., Perrigio, A., Antonelli, A., Eds.; Wiley-Blackwell: Chichester, UK, 2018; pp. 429–448. [Google Scholar]
- Rahbek, C.; Borregaard, M.K.; Antonelli, A.; Colwell, R.K.; Holt, B.G.; Nogues-Bravo, D.; Rasmussen, C.M.Ø.; Richardson, K.; Rosing, M.T.; Whittaker, R.J.; et al. Building mountain biodiversity: Geological and evolutionary processes. Science 2019, 365, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.N.; Ree, R.H.; Spicer, R.A.; Xing, Y.W. Ancient orogenic and monsoon-driven assembly of the world’s richest temperate alpine flora. Science 2020, 369, 578–581. [Google Scholar] [CrossRef]
- Ru, D.; Sun, Y.; Wang, D.; Chen, Y.; Wang, T.; Hu, Q.; Abbott, R.J.; Liu, J. Population genomic analysis reveals that homoploid hybrid speciation can be a lengthy process. Mol. Ecol. 2018, 27, 4875–4887. [Google Scholar] [CrossRef]
- Wu, S.; Wang, Y.; Wang, Z.; Shrestha, N.; Liu, J. Species divergence with gene flow and hybrid speciation on the Qinghai–Tibet Plateau. New Phytol. 2022, 234, 392–404. [Google Scholar] [CrossRef]
- Liu, J.; Lindstrom, A.J.; Gong, Y.; Dong, S.; Liu, Y.C.; Zhang, S.; Gong, X. Eco-evolutionary evidence for the global diversity pattern of Cycas (Cycadaceae). J. Integr. Plant Biol. 2024, 66, 1170–1191. [Google Scholar] [CrossRef]
- Huang, D.Q.; Ma, X.G.; Sun, H. Phylogenomic analyses and chromosome ploidy identification reveal multiple cryptic species in Allium sikkimense complex (Amaryllidaceae). Front. Plant Sci. 2024, 14, 1268546. [Google Scholar] [CrossRef]
- Wu, L.L.; Cui, X.K.; Milne, R.I.; Sun, Y.S.; Liu, J.Q. Multiple autopolyploidizations and range expansion of Allium przewalskianum Regel. (Alliaceae) in the Qinghai-Tibetan Plateau. Mol. Ecol. 2010, 19, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Van Drunen, W.E.; Husband, B.C. Evolutionary associations between polyploidy, clonal reproduction, and perenniality in the angiosperms. New Phytol. 2019, 224, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Zheng, Q.J.; Onstein, R.E.; Rojas-Andres, B.M.; Hauenschild, F.; Muellner-Riehl, A.N.; Xing, Y.W. Polyploidy promotes species diversification of Allium through ecological shifts. New Phytol. 2020, 225, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 1–14. [Google Scholar] [CrossRef]
- Löytynoja, A.; Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 2008, 320, 1632–1635. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef]
- Sayyari, E.; Mirarab, S. Fast Coalescent-Based Computation of Local Branch Support from Quartet Frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef]
- Smith, S.A.; Moore, M.J.; Brown, J.W.; Hinchliff, C.E. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 2015, 15, 150. [Google Scholar] [CrossRef]
- Salichos, L.; Stamatakis, A.; Rokas, A. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 2014, 31, 1261–1271. [Google Scholar] [CrossRef]
- Doyle, J.J. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef]
- Pease, J.B.; Brown, J.W.; Walker, J.F.; Hinchliff, C.E.; Smith, S.A. Quartet Sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 2018, 105, 385–403. [Google Scholar] [CrossRef]
- Allman, E.S.; Mitchell, J.D.; Rhodes, J.A. Gene tree discord, simplex plots, and statistical tests under the coalescent. Syst. Biol. 2022, 71, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.A.; Baños, H.; Mitchell, J.D.; Allman, E.S. MSCquartets 1.0: Quartet methods for species trees and networks under the multispecies coalescent model in R. Bioinformatics 2021, 37, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.Y.; Jia, K.H.; Li, N.W.; Zhou, M.J.; Yang, H.; Tian, X.L.; Ma, P.F.; Zhang, R.G. Phytop: A tool for visualizing and recognizing signals of incomplete lineage sorting and hybridization using species trees output from ASTRAL. Hortic. Res. 2024, 11, uhae330. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Nakhleh, L. A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 2015, 16, S10. [Google Scholar] [CrossRef]
- Solís-Lemus, C.; Bastide, P.; Ané, C. PhyloNetworks: A package for phylogenetic networks. Mol. Biol. Evol. 2017, 34, 3292–3298. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef]
- Allman, E.S.; Baños, H.; Rhodes, J.A. NANUQ: A method for inferring species networks from gene trees under the coalescent model. Algorithms Mol. Biol. 2019, 14, 1–25. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Allium cyathophorum | Allium tetraploideum | Allium farreri | Allium siphonanthum | Allium spicatum | Allium mairei |
|---|---|---|---|---|---|---|
| bulb | solitary or clustered, cylindric | solitary or clustered, cylindric | solitary or clustered, cylindric | solitary or clustered, cylindric | solitary, cylindric | usually clustered, cylindric |
| bulb tunics | grayish brown, fibrous | grayish brown, fibrous | grayish brown, fibrous | yellowish brown, subreticulate | yellowish brown, fibrous | yellowish brown to grayish brown, fibrous |
| scape shape | terete, usually 2-angled, covered with leaf sheaths only at base | terete, usually 3-angled, covered with leaf sheaths only at base | terete, usually 2-angled, covered with leaf sheaths only at base | terete, not angled, covered with leaf sheaths only at base | terete, hollow, not angled, covered with leaf sheaths only at base | terete, 2-angled, covered with leaf sheaths only at base. |
| scape length | 13–25 cm | 13–25 cm | 13–25 cm | 18–60 cm | 5–40 cm | 10–30 cm |
| leaf | shorter than scape | shorter than scape | shorter than scape | subequal to scape | subequal to scape | shorter than or subequal to scape |
| inflorescence | umbel hemispheric, laxly flowered | umbel hemispheric, laxly flowered | umbel hemispheric, laxly flowered | umbel globose, densely many flowered | spike, densely many flowered | umbel, with very few flowers |
| pedicel length | equal, 1–3 × as long as perianth, ebracteolate | equal, 1–3 × as long as perianth, ebracteolate | equal, 1–3 × as long as perianth, ebracteolate | equal, shorter than 1/3 of perianth, ebracteolate | equal, shorter than 1/3 of perianth, ebracteolate | unequal, 1.5–2 × as long as perianth, ebracteolate |
| spathe | 1(–3)-valved, persistent | 1(–3)-valved, persistent | 1(–3)-valved, persistent | 2-valved, persistent | 1-valved, persistent | 1-valved, persistent |
| perianth | purple, retuse to obtuse at apex | dark maroon, retuse to obtuse at apex | purple, acuminate at apex | white to purple-red, retuse to obtuse at apex | white to purple-red, retuse to obtuse at apex | pale red to purple-red, apex obtuse or acute |
| inner filaments | shoulder-shaped at base, no teeth | shoulder-shaped at base, no teeth | triangular at base, no teeth | wide at base, entire or 1-toothed on each side | wide at base, entire or 2-toothed on each side | conical at base, no teeth |
| style | shorter than ovary | shorter than ovary | shorter than ovary | longer than ovary | longer than ovary | shorter than ovary |
| chromosome number | 2n = 16 | 2n = 32 | 2n = 16 | unknown | 2n = 16 | 2n = 16, 32 |
| flowering season | June to August | June to August | June to August | September to October | August to October | August to October |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Tang, Z.-J.; Wang, Y.; Tan, J.-B.; Zhou, S.-D.; He, X.-J.; Xie, D.-F. Phylogenomic Analyses Reveal Species Relationships and Phylogenetic Incongruence with New Member Detected in Allium Subgenus Cyathophora. Plants 2025, 14, 2083. https://doi.org/10.3390/plants14132083
Chen K, Tang Z-J, Wang Y, Tan J-B, Zhou S-D, He X-J, Xie D-F. Phylogenomic Analyses Reveal Species Relationships and Phylogenetic Incongruence with New Member Detected in Allium Subgenus Cyathophora. Plants. 2025; 14(13):2083. https://doi.org/10.3390/plants14132083
Chicago/Turabian StyleChen, Kun, Zi-Jun Tang, Yuan Wang, Jin-Bo Tan, Song-Dong Zhou, Xing-Jin He, and Deng-Feng Xie. 2025. "Phylogenomic Analyses Reveal Species Relationships and Phylogenetic Incongruence with New Member Detected in Allium Subgenus Cyathophora" Plants 14, no. 13: 2083. https://doi.org/10.3390/plants14132083
APA StyleChen, K., Tang, Z.-J., Wang, Y., Tan, J.-B., Zhou, S.-D., He, X.-J., & Xie, D.-F. (2025). Phylogenomic Analyses Reveal Species Relationships and Phylogenetic Incongruence with New Member Detected in Allium Subgenus Cyathophora. Plants, 14(13), 2083. https://doi.org/10.3390/plants14132083