
Exploring the Genetic Networks of HLB Tolerance in Citrus: Insights Across Species and Tissues

 , , , ,
, , , ,  ,
,  and
and
Abstract
1. Introduction
2. Results
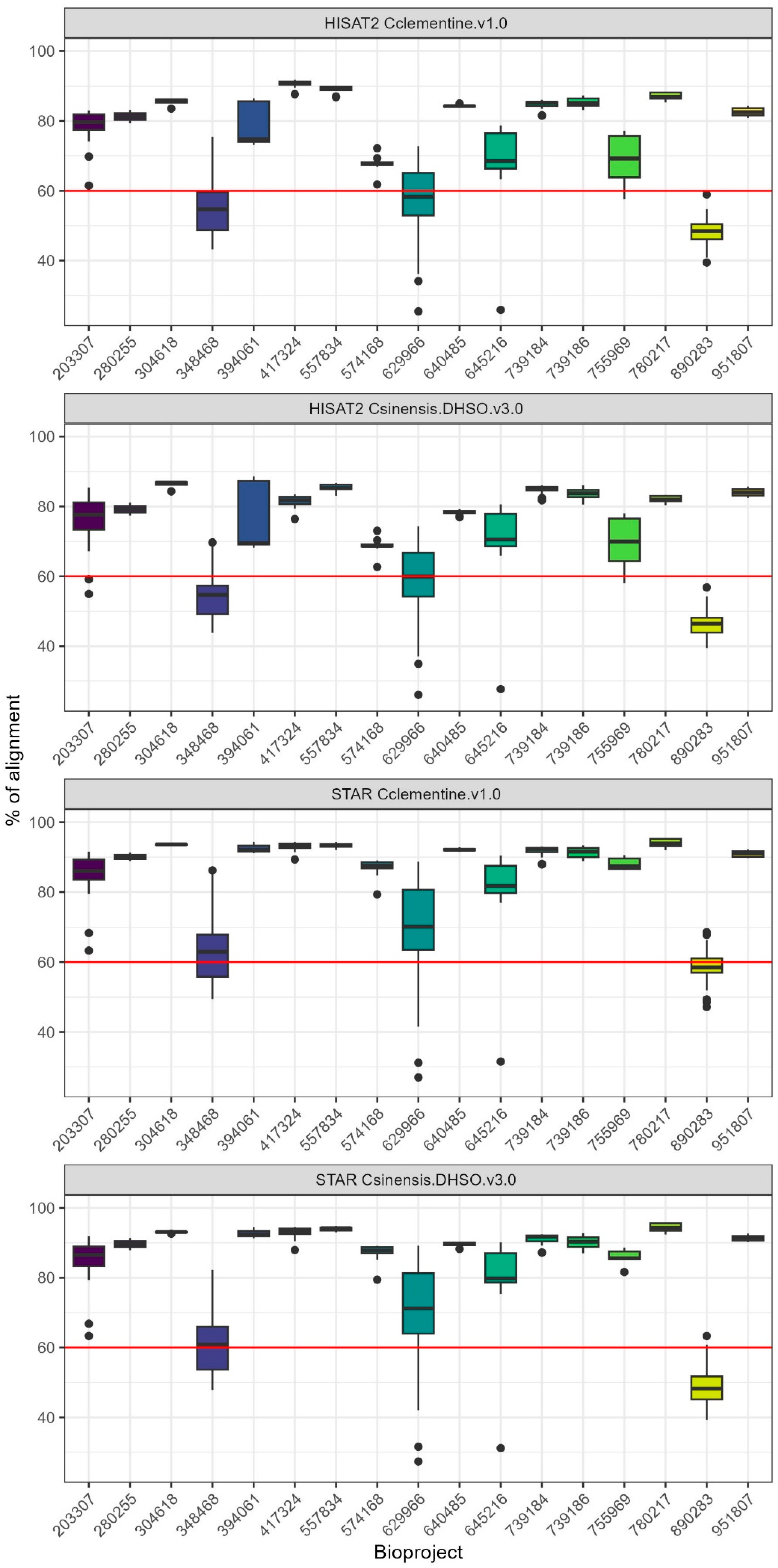
2.1. Evaluation of RNA-Seq Aligners and Selection of Citrus Reference Genome for Optimal Mapping Efficiency
2.2. Quality Control and Data Retention for Further Analyses
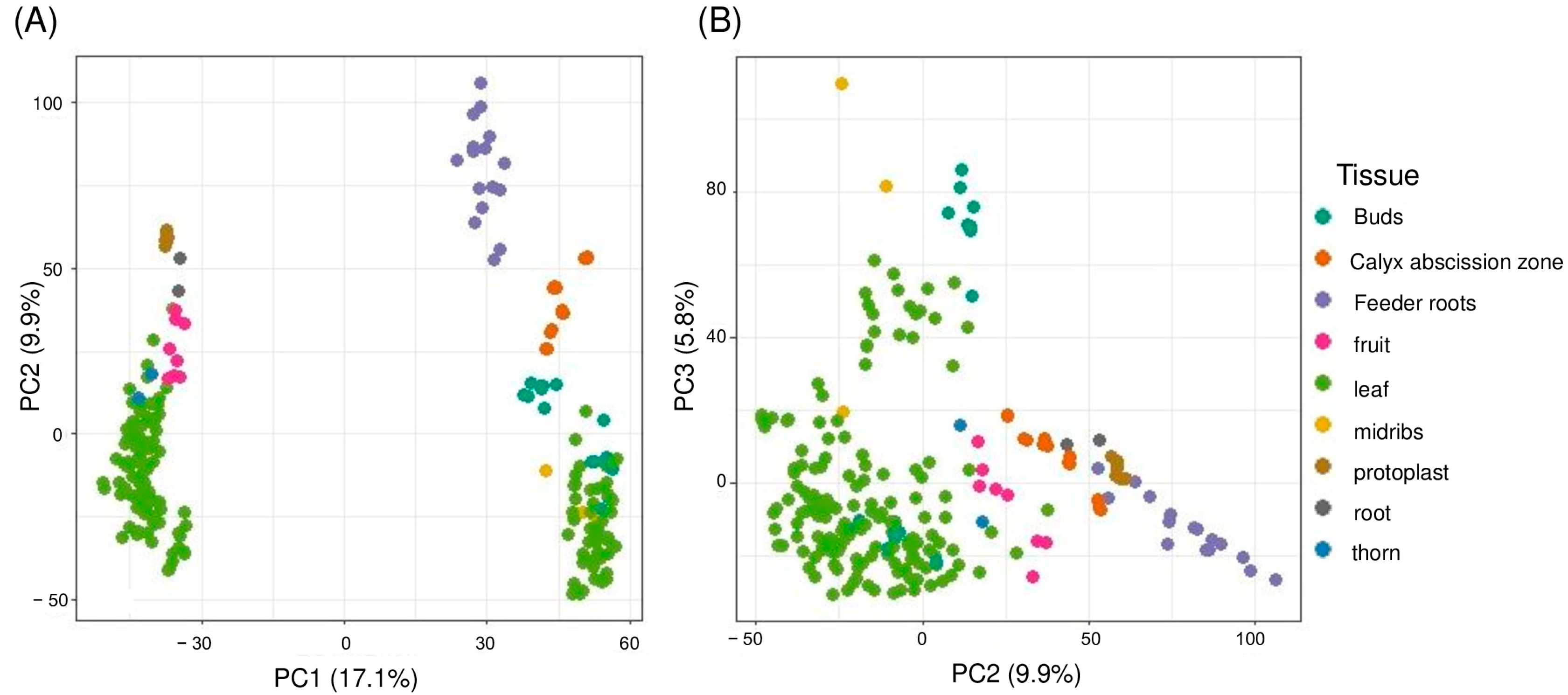
2.3. Gene Expression Profiling and Principal Component Analysis
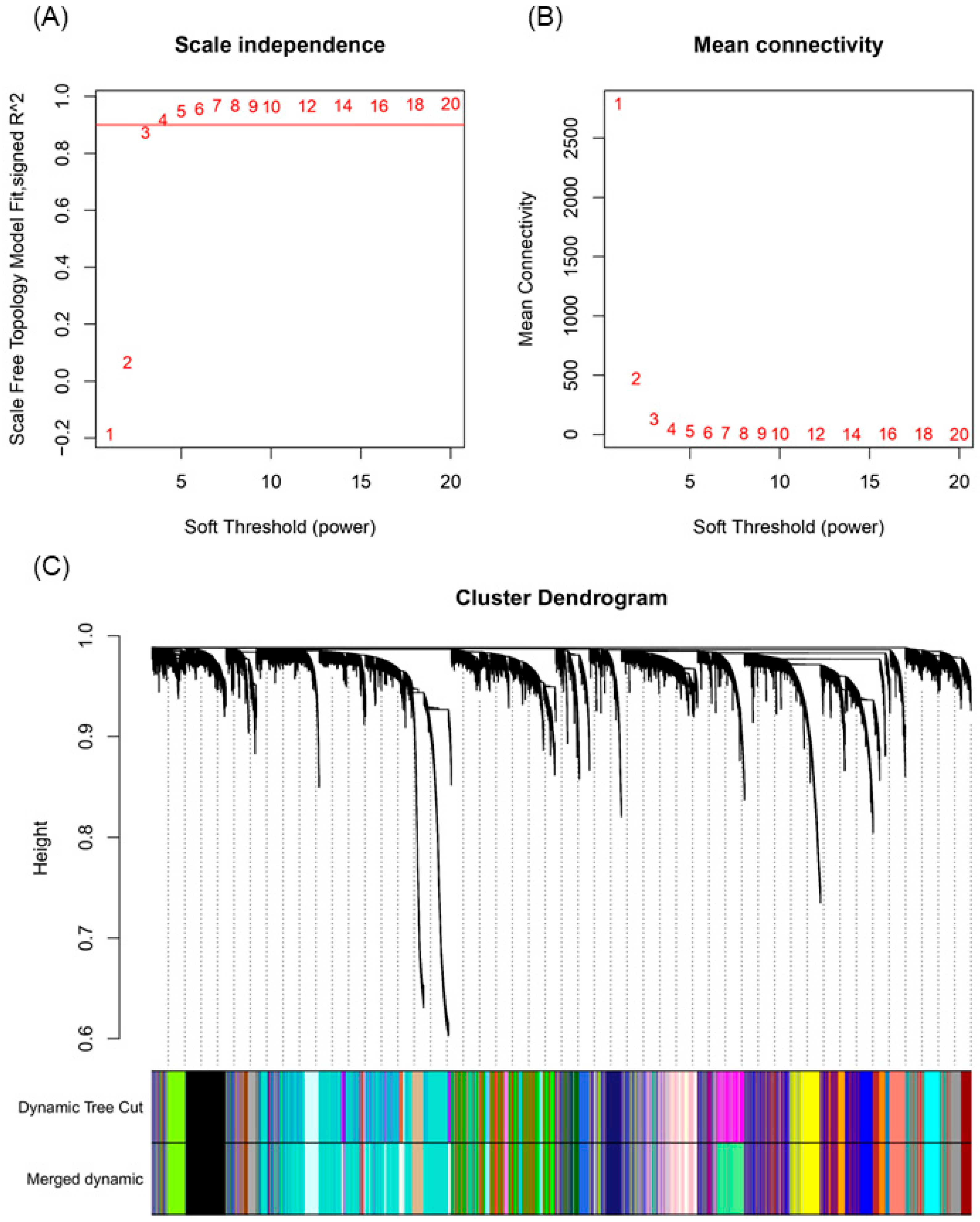
2.4. Clustering of Gene Expression Samples
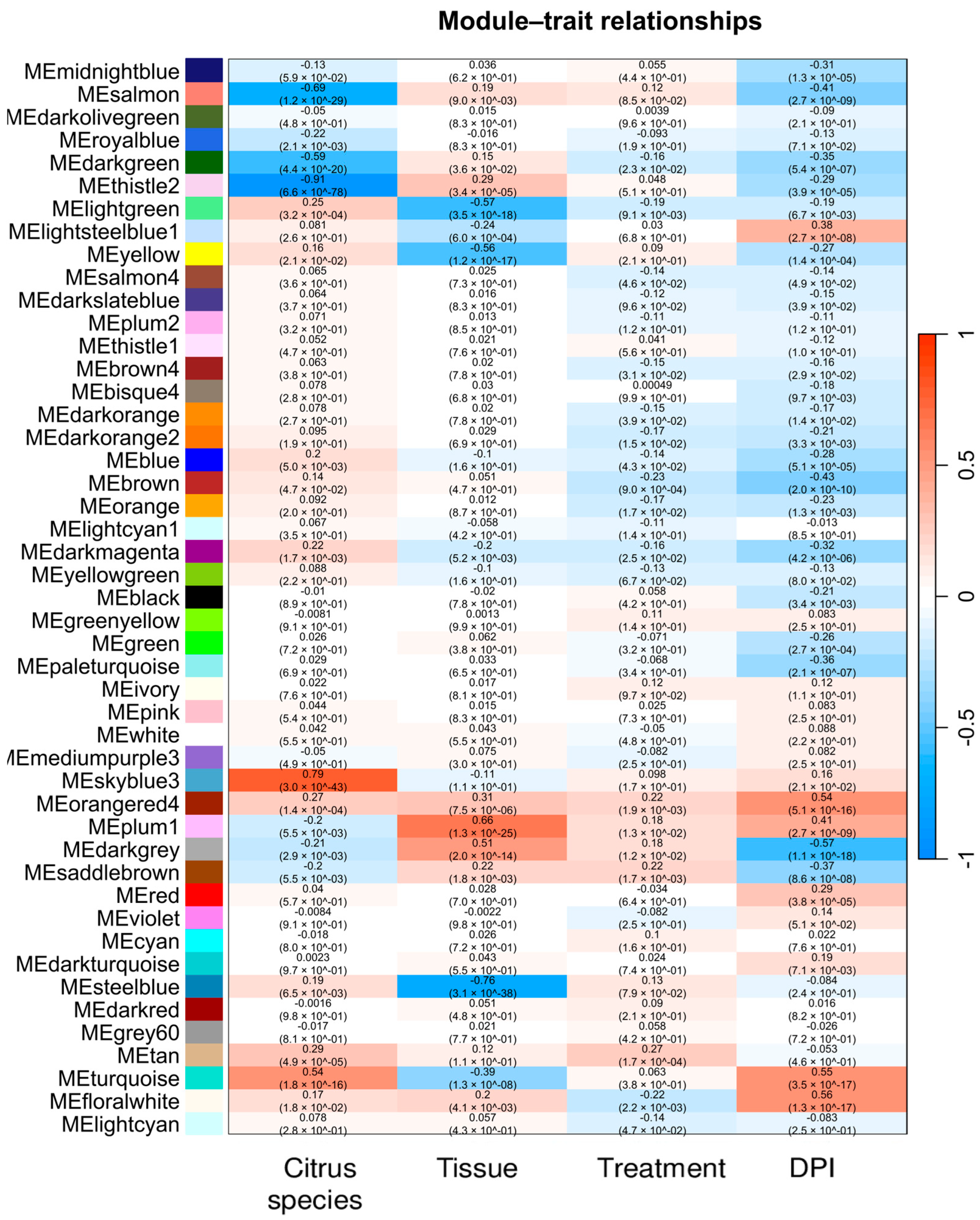
2.5. Weighted Co-Expression Network Construction and Module Trait Analysis
2.6. Gene Significance and Module Membership Results
2.7. Differential Gene Expression Analysis Across Selected Citrus Bioprojects
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. Quality Control and Mapping of Data
4.3. Data Normalization and Outlier Detection
4.4. Weighted Gene Co-Expression Network Analysis
4.5. Gene Significance (GS) and Module Membership (MM) Evaluation
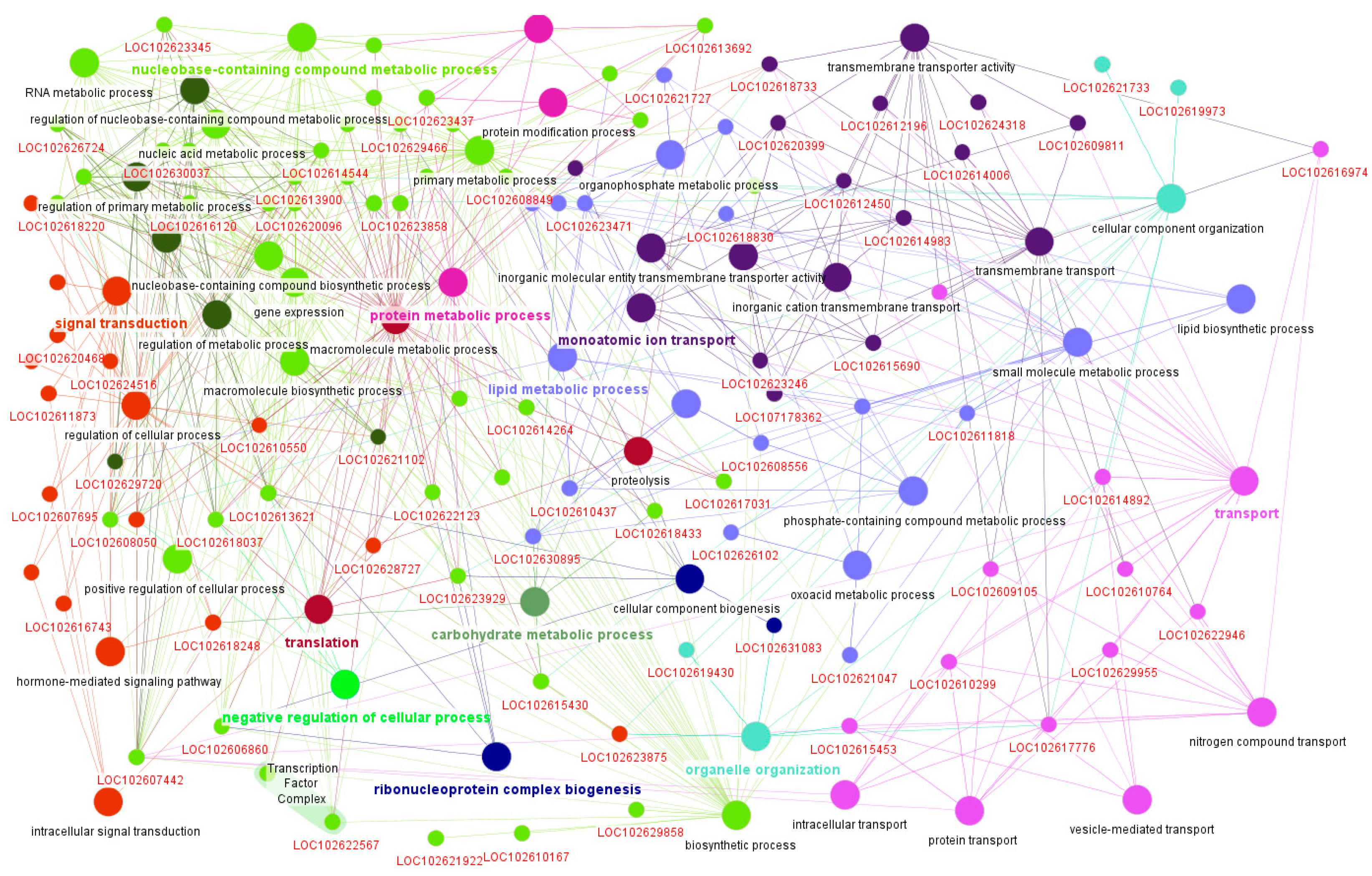
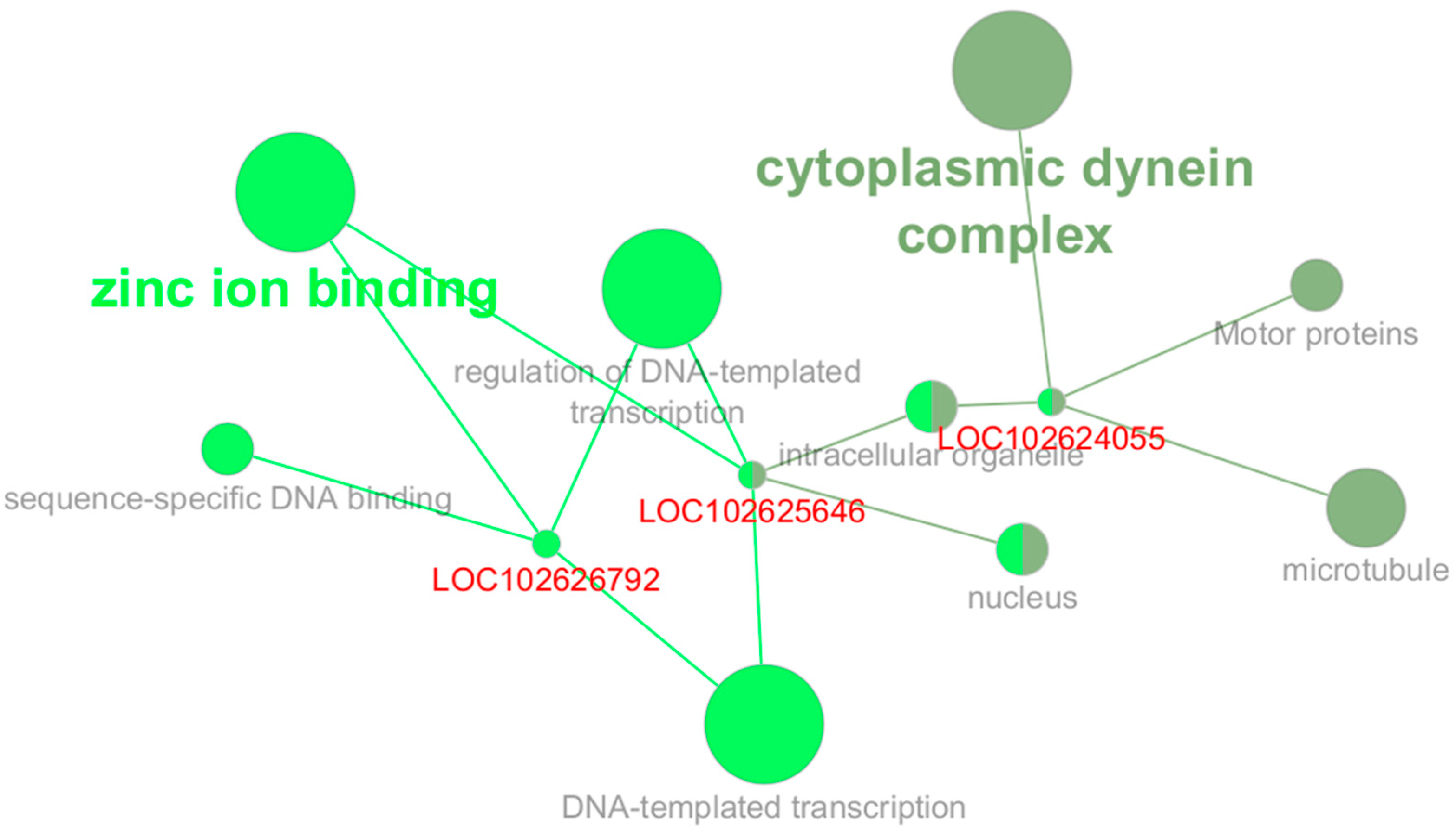
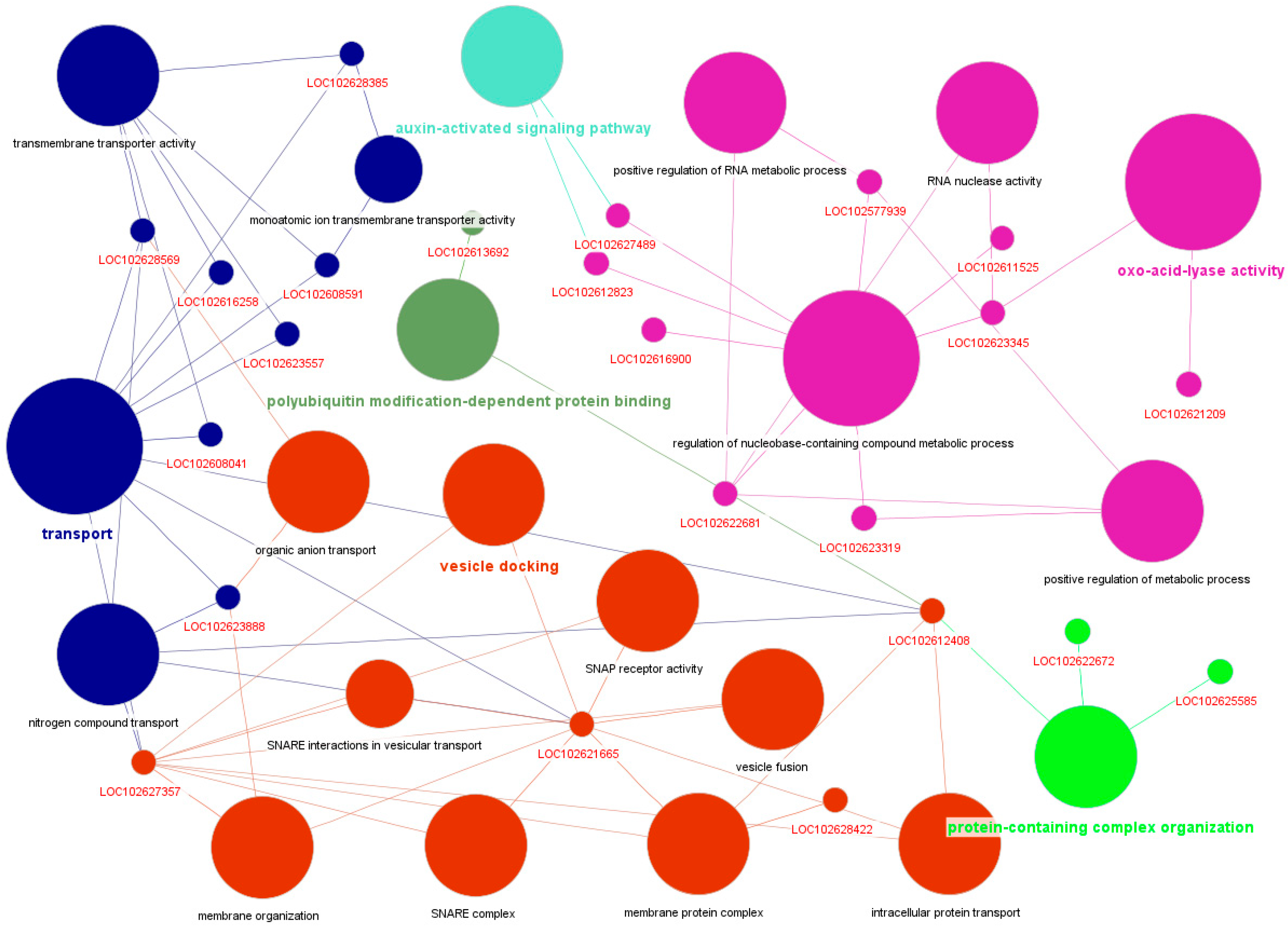
4.6. Functional Annotation and Network Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bové, J.M. Huanglongbing: A Destructive, Newly-Emerging, Century-Old Disease of Citrus. J. Plant Pathol. 2006, 88, 7–37. [Google Scholar]
- Gottwald, T.R. Current Epidemiological Understanding of Citrus Huanglongbing. Annu. Rev. Phytopathol. 2010, 48, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Kokane, S.; Savita, B.K.; Kumar, P.; Sharma, A.K.; Ozcan, A.; Kokane, A.; Santra, S. Huanglongbing Pandemic: Current Challenges and Emerging Management Strategies. Plants 2023, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Reinking, O.A. Diseases of Economic Plants in Southern China. Philipp. Agric. 1919, 8, 109–134. [Google Scholar]
- Husain, M.A.; Nath, D. The Citrus Psylla (Diaphorina Citri, Kuw.) Psyllidae: Homoptera. In Memoirs of Department of Agriculture in India (Entomological Series); Thacker, Spink, and Company: Calcutta, India, 1927; Volume 10, pp. 5–27. [Google Scholar]
- Capoor, S.P. Decline of Citrus Trees in India. Bull. Natl. Inst. Sci. India 1963, 24, 48–64. [Google Scholar]
- Van der Merwe, A.J.; Anderssen, F.G. Chromium and Manganese Toxicity. Is It Important in Transvaal Citrus Growing? Farm 1937, 12, 439–440. [Google Scholar]
- Boa, E. Citrus Huanglongbing (Greening) Disease. Plant Health Cases 2023, 2023, 0003. [Google Scholar] [CrossRef]
- Coletta-Filho, H.D.; Targon, M.L.P.N.; Takita, M.A.; De Negri, J.D.; Pompeu, J.; Machado, M.A.; do Amaral, A.M.; Muller, G.W. First Report of the Causal Agent of Huanglongbing (“Candidatus Liberibacter Asiaticus”) in Brazil. Plant Dis. 2004, 88, 1382. [Google Scholar] [CrossRef]
- Halbert, S.E. The Discovery of Huanglongbing in Florida. In Proceedings of the International Citrus Canker and Huanglongbing Research Workshop, Orlando, FL, USA, 7–11 November 2005. [Google Scholar]
- European and Mediterranean Plant Protection Organization ‘Candidatus Liberibacter Asiaticus’. EPPO Datasheets on Pests Recommended for Regulation. Available online: https://gd.eppo.int/taxon/LIBEAS/distribution (accessed on 14 September 2024).
- Usman, H.M.; Saleem, U.; Shafique, T. Decoding Huanglongbing: Understanding the Origins, Impacts, and Remedies for Citrus Greening. Phytopathogenomics Dis. Control. 2023, 2, 13–20. [Google Scholar] [CrossRef]
- Dala-Paula, B.M.; Plotto, A.; Bai, J.; Manthey, J.A.; Baldwin, E.A.; Ferrarezi, R.S.; Gloria, M.B.A. Effect of Huanglongbing or Greening Disease on Orange Juice Quality, a Review. Front. Plant Sci 2019, 9, 1976. [Google Scholar] [CrossRef]
- Weber, K.C.; Mahmoud, L.M.; Stanton, D.; Welker, S.; Qiu, W.; Grosser, J.W.; Levy, A.; Dutt, M. Insights into the Mechanism of Huanglongbing Tolerance in the Australian Finger Lime (Citrus australasica). Front. Plant Sci. 2022, 13, 1019295. [Google Scholar] [CrossRef]
- Peng, Z.; Bredeson, J.V.; Wu, G.A.; Shu, S.; Rawat, N.; Du, D.; Parajuli, S.; Yu, Q.; You, Q.; Rokhsar, D.S.; et al. A Chromosome-Scale Reference Genome of Trifoliate Orange (Poncirus Trifoliata) Provides Insights into Disease Resistance, Cold Tolerance and Genome Evolution in Citrus. Plant J. 2020, 104, 1215–1232. [Google Scholar] [CrossRef] [PubMed]
- Killiny, N.; Jones, S.E. Metabolic Alterations in the Nymphal Instars of Diaphorina Citri Induced by Candidatus Liberibacter Asiaticus, the Putative Pathogen of Huanglongbing. PLoS ONE 2018, 13, e0191871. [Google Scholar] [CrossRef]
- Wu, H.; Hu, Y.; Fu, S.; Zhou, C.; Wang, X. Coordination of Multiple Regulation Pathways Contributes to the Tolerance of a Wild Citrus Species (Citrus ichangensis ‘2586’) against Huanglongbing. Physiol. Mol. Plant Pathol. 2020, 109, 101457. [Google Scholar] [CrossRef]
- Folimonova, S.Y.; Robertson, C.J.; Garnsey, S.M.; Gowda, S.; Dawson, W.O. Examination of the Responses of Different Genotypes of Citrus to Huanglongbing (Citrus greening) under Different Conditions. Phytopathology 2009, 99, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Bai, X.; Wen, Q.; Xie, Z.; Wu, L.; Peng, A.; He, Y.; Xu, L.; Chen, S. Comparative Analysis of Tolerant and Susceptible Citrus Reveals the Role of Methyl Salicylate Signaling in the Response to Huanglongbing. J. Plant Growth Regul. 2019, 38, 1516–1528. [Google Scholar] [CrossRef]
- Hu, Y.; Zhong, X.; Liu, X.; Lou, B.; Zhou, C.; Wang, X. Comparative Transcriptome Analysis Unveils the Tolerance Mechanisms of Citrus Hystrix in Response to “Candidatus Liberibacter Asiaticus” Infection. PLoS ONE 2017, 12, e0189229. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, L.; Yu, X.; Stover, E.; Luo, F.; Duan, Y. Transcriptome Profiling of Huanglongbing (HLB) Tolerant and Susceptible Citrus Plants Reveals the Role of Basal Resistance in HLB Tolerance. Front. Plant Sci. 2016, 7, 933. [Google Scholar] [CrossRef]
- Ramadugu, C.; Keremane, M.L.; Halbert, S.E.; Duan, Y.P.; Roose, M.L.; Stover, E.; Lee, R.F. Long-Term Field Evaluation Reveals Huanglongbing Resistance in Citrus Relatives. Plant Dis. 2016, 100, 1858–1869. [Google Scholar] [CrossRef]
- Miles, G.P.; Stover, E.; Ramadugu, C.; Keremane, M.L.; Lee, R.F. Apparent Tolerance to Huanglongbing in Citrus and Citrus-Related Germplasm. HortScience 2017, 52, 31–39. [Google Scholar] [CrossRef]
- Alves, M.N.; Lopes, S.A.; Raiol-Junior, L.L.; Wulff, N.A.; Girardi, E.A.; Ollitrault, P.; Peña, L. Resistance to ‘Candidatus Liberibacter Asiaticus,’ the Huanglongbing Associated Bacterium, in Sexually and/or Graft-Compatible Citrus Relatives. Front. Plant Sci. 2021, 11, 617664. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Niu, D.D.; Kund, G.; Jones, M.; Albrecht, U.; Nguyen, L.; Bui, C.; Ramadugu, C.; Bowman, K.D.; Trumble, J.; et al. Identification of Citrus Immune Regulators Involved in Defence against Huanglongbing Using a New Functional Screening System. Plant Biotechnol. J. 2021, 19, 757–766. [Google Scholar] [CrossRef]
- Sivager, G.; Calvez, L.; Bruyere, S.; Boisne-Noc, R.; Brat, P.; Gros, O.; Ollitrault, P.; Morillon, R. Specific Physiological and Anatomical Traits Associated with Polyploidy and Better Detoxification Processes Contribute to Improved Huanglongbing Tolerance of the Persian Lime Compared with the Mexican Lime. Front. Plant Sci. 2021, 12, 685679. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.P.; De Francesco, A.; Trinh, J.; Gurung, F.B.; Pang, Z.; Vidalakis, G.; Wang, N.; Ancona, V.; Ma, W.; Coaker, G. Genome-Wide Analyses of Liberibacter Species Provides Insights into Evolution, Phylogenetic Relationships, and Virulence Factors. Mol. Plant Pathol. 2020, 21, 716–731. [Google Scholar] [CrossRef]
- Batarseh, T.N.; Batarseh, S.N.; Morales-Cruz, A.; Gaut, B.S. Comparative Genomics of the Liberibacter Genus Reveals Widespread Diversity in Genomic Content and Positive Selection History. Front. Microbiol. 2023, 14, 1206094. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Pierson, E.A.; Setubal, J.C.; Xu, J.; Levy, J.G.; Zhang, Y.; Li, J.; Rangel, L.T.; Martins, J. The Candidatus Liberibacter-Host Interface: Insights into Pathogenesis Mechanisms and Disease Control. Annu. Rev. Phytopathol. 2017, 55, 451–482. [Google Scholar] [CrossRef]
- Guo, C.F.; Kong, W.Z.; Mukangango, M.; Hu, Y.W.; Liu, Y.T.; Sang, W.; Qiu, B.L. Distribution and Dynamic Changes of Huanglongbing Pathogen in Its Insect Vector Diaphorina Citri. Front. Cell. Infect. Microbiol. 2024, 14, 1408362. [Google Scholar] [CrossRef]
- Endarto, O.; Wicaksono, R.C.; Wuryantini, S.; Tarno, H.; Nurindah. Climate Change Mitigation and Seasonal Infestation Patterns of Citrus Psyllid Diaphorina Citri: Implications for Managing Huanglongbing (HLB) Disease in Tangerine Citrus. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Padang, Indonesia, 1 October 2023; Institute of Physics: London, UK, 2024; Volume 1346. [Google Scholar]
- Pierson, E.A.; Cubero, J.; Roper, C.; Brown, J.K.; Bock, C.H.; Wang, N. ‘Candidatus Liberibacter’ Pathosystems at the Forefront of Agricultural and Biological Research Challenges. Phytopathology 2022, 112, 7–10. [Google Scholar] [CrossRef]
- Yao, L.; Guo, X.; Su, J.; Zhang, Q.; Lian, M.; Xue, H.; Li, Q.; He, Y.; Zou, X.; Song, Z.; et al. ABA-CsABI5-CsCalS11 Module Upregulates Callose Deposition of Citrus Infected with Candidatus Liberibacter Asiaticus. Hortic. Res. 2024, 11, uhad276. [Google Scholar] [CrossRef]
- Basu, S.; Huynh, L.; Zhang, S.; Rabara, R.; Nguyen, H.; Velásquez Guzmán, J.; Hao, G.; Miles, G.; Shi, Q.; Stover, E.; et al. Two Liberibacter Proteins Combine to Suppress Critical Innate Immune Defenses in Citrus. Front. Plant Sci. 2022, 13, 869178. [Google Scholar] [CrossRef]
- Pandey, S.S.; Xu, J.; Achor, D.S.; Li, J.; Wang, N. Microscopic and Transcriptomic Analyses of Early Events Triggered by ‘Candidatus Liberibacter Asiaticus’ in Young Flushes of Huanglongbing-Positive Citrus Trees. Phytopathology 2023, 113, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, C.; Turner, D.; Wang, C.; Welker, S.; Achor, D.; Artiga, Y.A.; Turgeon, R.; Levy, A. Candidatus Liberibacter Asiaticus Reduces Callose and reactive Oxygen Species Production in the Phloem. bioRxiv 2022. [Google Scholar]
- Shi, J.; Gong, Y.; Shi, H.; Ma, X.; Zhu, Y.; Yang, F.; Wang, D.; Fu, Y.; Lin, Y.; Yang, N.; et al. ‘Candidatus Liberibacter Asiaticus’ Secretory Protein SDE3 Inhibits Host Autophagy to Promote Huanglongbing Disease in Citrus. Autophagy 2023, 19, 2558–2574. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Liu, L.; Fox, E.G.P.; Deng, X.; Xu, M.; Zheng, Z.; Li, X.; Fu, J.; Zhu, H.; Huang, J.; et al. Physiological Variables Influenced by ‘Candidatus Liberibacter Asiaticus’ Infection in Two Citrus Species. Plant Dis. 2023, 107, 1769–1776. [Google Scholar] [CrossRef]
- Fan, J.; Chen, C.; Yu, Q.; Khalaf, A.; Achor, D.S.; Brlansky, R.H.; Moore, G.A.; Li, Z.G.; Gmitter, F.G. Comparative Transcriptional and Anatomical Analyses of Tolerant Rough Lemon and Susceptible Sweet Orange in Response to “Candidatus Liberibacter Asiaticus” Infection. Mol. Plant-Microbe Interact. 2012, 25, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Chen, C.; Du, D.; Huang, M.; Yao, J.; Yu, F.; Brlansky, R.H.; Gmitter, F.G. Reprogramming of a Defense Signaling Pathway in Rough Lemon and Sweet Orange Is a Critical Element of the Early Response to Candidatus Liberibacter Asiaticus. Hortic. Res. 2017, 4, 17063. [Google Scholar] [CrossRef]
- Ribeiro, C.; Xu, J.; Hendrich, C.; Pandey, S.S.; Yu, Q.; Gmitter, F.G., Jr.; Wang, N. Seasonal Transcriptome Profiling of Susceptible and Tolerant Citrus Cultivars to Citrus Huanglongbing. Phytopathology 2023, 113, 286–298. [Google Scholar] [CrossRef]
- Zhuo, X.; Yu, Q.; Russo, R.; Zhang, Y.; Wei, X.; Wang, Y.Z.; Holden, P.M.; Gmitter, F.G. Role of Long Non-Coding RNA in Regulatory Network Response to Candidatus Liberibacter Asiaticus in Citrus. Front. Plant Sci. 2023, 14, 1090711. [Google Scholar] [CrossRef]
- Wei, X.; Mira, A.; Yu, Q.; Gmitter, F.G. The Mechanism of Citrus Host Defense Response Repression at Early Stages of Infection by Feeding of Diaphorina Citri Transmitting Candidatus Liberibacter Asiaticus. Front. Plant Sci. 2021, 12, 1090711. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, K.; Chen, D.; Liu, K.; Chen, W.; He, F.; Tong, Z.; Luo, Q. Co-Expression Network Analysis and Identification of Core Genes in the Interaction between Wheat and Puccinia striiformis f. Sp. Tritici. Arch. Microbiol. 2023, 206, 241. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, P.; Fang, H.; Zheng, J.; Zhong, C.; Yang, Y.; Yu, W. Weighted Gene Co-Expression Analysis Network-Based Analysis on the Candidate Pathways and Hub Genes in Eggplant Bacterial Wilt-Resistance: A Plant Research Study. Int. J. Mol. Sci. 2021, 22, 13279. [Google Scholar] [CrossRef]
- Gao, P.; Wei, H.; Liu, J.; Chen, Z.; Qi, Y.; Wu, Z.; Huang, F.; Yu, L. Weighted Gene Coexpression Network Analysis of Candidate Pathways and Genes in Soft Rot Resistance of Amorphophallus. J. Am. Soc. Hortic. Sci. 2022, 147, 322–333. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; Hu, Y.; Huang, G. Analysis of Huanglongbing-Associated RNA-Seq Data Reveals Disturbances in Biological Processes within Citrus spp. Triggered by Candidatus Liberibacter Asiaticus Infection. Front. Plant Sci. 2024, 15, 1388163. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Di Marco, A.; Pellegrini, C. Comparing HISAT and STAR-Based Pipelines for RNA-Seq Data Analysis: A Real Experience. In Proceedings of the IEEE Symposium on Computer-Based Medical Systems, L’Aquila, Italy, 22–24 June 2023; Institute of Electrical and Electronics Engineers Inc.: Piscataway Township, NJ, USA, 2023; Volume 2023, pp. 218–224. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Martin, L.B.B.; Fei, Z.; Giovannoni, J.J.; Rose, J.K.C. Catalyzing Plant Science Research with RNA-Seq. Front. Plant Sci. 2013, 4, 66. [Google Scholar] [CrossRef]
- Martinelli, F.; Uratsu, S.L.; Albrecht, U.; Reagan, R.L.; Phu, M.L.; Britton, M.; Buffalo, V.; Fass, J.; Leicht, E.; Zhao, W.; et al. Transcriptome Profiling of Citrus Fruit Response to Huanglongbing Disease. PLoS ONE 2012, 7, e38039. [Google Scholar] [CrossRef]
- van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene Co-Expression Analysis for Functional Classification and Gene-Disease Predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Balan, B.; Ibáñez, A.M.; Dandekar, A.M.; Caruso, T.; Martinelli, F. Identifying Host Molecular Features Strongly Linked with Responses to Huanglongbing Disease in Citrus Leaves. Front. Plant Sci. 2018, 9, 277. [Google Scholar] [CrossRef]
- Thakuria, D.; Chaliha, C.; Dutta, P.; Sinha, S.; Uzir, P.; Singh, S.B.; Hazarika, S.; Sahoo, L.; Kharbikar, L.L.; Singh, D. Citrus Huanglongbing (HLB): Diagnostic and Management Options. Physiol. Mol. Plant Pathol. 2023, 125, 102016. [Google Scholar] [CrossRef]
- Wei, X.; Moreno-Hagelsieb, G.; Glick, B.R.; Doxey, A.C. Comparative Analysis of Adenylate Isopentenyl Transferase Genes in Plant Growth-Promoting Bacteria and Plant Pathogenic Bacteria. Heliyon 2023, 9, e13955. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Tang, X.; Zhang, Y.; Gmitter, F.G.; Wang, Y. Metabolomic Analysis Provides New Insight Into Tolerance of Huanglongbing in Citrus. Front. Plant Sci. 2021, 12, 710598. [Google Scholar] [CrossRef]
- Kwakye, S.; Kadyampakeni, D.M. Micronutrients Improve Growth and Development of HLB-Affected Citrus Trees in Florida. Plants 2023, 12, 73. [Google Scholar] [CrossRef]
- Masaoka, Y.; Pustika, A.; Subandiyah, S.; Okada, A.; Hanundin, E.; Purwanto, B.; Okuda, M.; Okada, Y.; Saito, A.; Holford, P.; et al. Lower Concentrations of Microelements in Leaves of Citrus Infected with ‘Candidatus Liberibacter Asiaticus’. Jpn. Agric. Res. Q. 2011, 45, 269–275. [Google Scholar] [CrossRef]
- Hamido, S.A.; Morgan, K.T.; Kadyampakeni, D.M. The Effect of Huanglongbing on Young Citrus Tree Water Use. Horttechnology 2017, 27, 659–665. [Google Scholar] [CrossRef]
- Graham, J.H.; Johnson, E.G.; Gottwald, T.R.; Irey, M.S. Presymptomatic Fibrous Root Decline in Citrus Trees Caused by Huanglongbing and Potential Interaction with Phytophthora spp. Plant Dis. 2013, 97, 1195–1199. [Google Scholar] [CrossRef]
- Killiny, N.; Nehela, Y. Metabolomic Response to Huanglongbing: Role of Carboxylic Compounds in Citrus Sinensis Response to “Candidatus Liberibacter Asiaticus” and Its Vector, Diaphorina Citri. Mol. Plant-Microbe Interact. 2017, 30, 666–678. [Google Scholar] [CrossRef]
- Gao, C.; Li, C.; Li, Z.; Liu, Y.; Li, J.; Guo, J.; Mao, J.; Fang, F.; Wang, C.; Deng, X.; et al. Comparative Transcriptome Profiling of Susceptible and Tolerant Citrus Species at Early and Late Stage of Infection by “Candidatus Liberibacter Asiaticus”. Front. Plant Sci. 2023, 14, 1191029. [Google Scholar] [CrossRef]
- Hu, B.; Rao, M.J.; Deng, X.; Pandey, S.S.; Hendrich, C.; Ding, F.; Wang, N.; Xu, Q. Molecular Signatures between Citrus and Candidatus Liberibacter Asiaticus. PLoS Pathog. 2021, 17, e1010071. [Google Scholar] [CrossRef]
- Bojórquez-Orozco, A.M.; Arce-Leal, Á.P.; Montes, R.A.C.; Santos-Cervantes, M.E.; Cruz-Mendívil, A.; Méndez-Lozano, J.; Castillo, A.G.; Rodríguez-Negrete, E.A.; Leyva-López, N.E. Differential Expression of MiRNAs Involved in Response to Candidatus Liberibacter Asiaticus Infection in Mexican Lime at Early and Late Stages of Huanglongbing Disease. Plants 2023, 12, 1039. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhong, Y.; Jiang, B.; Yan, H.; Ren, S.; Cheng, C. MicroRNA MiR171b Positively Regulates Resistance to Huanglongbing of Citrus. Int. J. Mol. Sci. 2023, 24, 5737. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Jeong, D.H.; de Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; González, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as Master Regulators of the Plant NB-LRR Defense Gene Family via the Production of Phased, Trans-Acting SiRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef] [PubMed]
- Pitino, M.; Armstrong, C.M.; Duan, Y. Molecular Mechanisms behind the Accumulation of ATP and H2O2 in Citrus Plants in Response to “Candidatus Liberibacter Asiaticus” Infection. Hortic. Res. 2017, 4, 17040. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, F.; Dandekar, A.M. Genetic Mechanisms of the Devious Intruder Candidatus Liberibacter in Citrus. Front. Plant Sci. 2017, 8, 904. [Google Scholar] [CrossRef]
- Albrecht, U.; Bowman, K.D. Transcriptional Response of Susceptible and Tolerant Citrus to Infection with Candidatus Liberibacter Asiaticus. Plant Sci. 2012, 185–186, 118–130. [Google Scholar] [CrossRef]
- Wu, B.; Yu, Q.; Deng, Z.; Duan, Y.; Luo, F.; Gmitter, F. A Chromosome-Level Phased Genome Enabling Allele-Level Studies in Sweet Orange: A Case Study on Citrus Huanglongbing Tolerance. Hortic. Res. 2023, 10, uhac247. [Google Scholar] [CrossRef]
- Deng, H.; Achor, D.; Exteberria, E.; Yu, Q.; Du, D.; Stanton, D.; Liang, G.; Gmitter, F.G. Phloem Regeneration Is a Mechanism for Huanglongbing-Tolerance of “Bearss” Lemon and “LB8-9” Sugar Belle® Mandarin. Front. Plant Sci. 2019, 10, 277. [Google Scholar] [CrossRef]
- Killiny, N.; Jones, S.E.; Nehela, Y.; Hijaz, F.; Dutt, M.; Gmitter, F.G.; Grosser, J.W. All Roads Lead to Rome: Towards Understanding Different Avenues of Tolerance to Huanglongbing in Citrus Cultivars. Plant Physiol. Biochem. 2018, 129, 1–10. [Google Scholar] [CrossRef]
- Martinelli, F.; Reagan, R.L.; Uratsu, S.L.; Phu, M.L.; Albrecht, U.; Zhao, W.; Davis, C.E.; Bowman, K.D.; Dandekar, A.M. Gene Regulatory Networks Elucidating Huanglongbing Disease Mechanisms. PLoS ONE 2013, 8, e74256. [Google Scholar] [CrossRef]
- Aritua, V.; Achor, D.; Gmitter, F.G.; Albrigo, G.; Wang, N. Transcriptional and Microscopic Analyses of Citrus Stem and Root Responses to Candidatus Liberibacter Asiaticus Infection. PLoS ONE 2013, 8, e73742. [Google Scholar] [CrossRef] [PubMed]
- Achor, D.; Welker, S.; Ben-Mahmoud, S.; Wang, C.; Folimonova, S.Y.; Dutt, M.; Gowda, S.; Levy, A. Dynamics of Candidatus Liberibacter Asiaticus Movement and Sieve-Pore Plugging in Citrus Sink Cells1[OPEN]. Plant Physiol. 2020, 182, 852–891. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, A.M.; Martinelli, F.; Reagan, R.L.; Uratsu, S.L.; Vo, A.; Tinoco, M.A.; Phu, M.L.; Chen, Y.; Rocke, D.M.; Dandekar, A.M. Transcriptome and Metabolome Analysis of Citrus Fruit to Elucidate Puffing Disorder. Plant Sci. 2014, 217–218, 87–98. [Google Scholar] [CrossRef]
- Zhong, Y.; Cheng, C.Z.; Jiang, N.H.; Jiang, B.; Zhang, Y.Y.; Wu, B.; Hu, M.L.; Zeng, J.W.; Yan, H.X.; Yi, G.J.; et al. Comparative Transcriptome and ITRAQ Proteome Analyses of Citrus Root Responses to Candidatus Liberibacter Asiaticus Infection. PLoS ONE 2015, 10, e0126973. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Shao, J.; Zhou, C.; Hartung, J.S. Transcriptome Analysis of Sweet Orange Trees Infected with “Candidatus Liberibacter Asiaticus” and Two Strains of Citrus Tristeza Virus. BMC Genom. 2016, 17, 349. [Google Scholar] [CrossRef]
- Fang, F.; Guo, H.; Zhao, A.; Li, T.; Liao, H.; Deng, X.; Xu, M.; Zheng, Z. A Significantly High Abundance of “Candidatus Liberibacter Asiaticus” in Citrus Fruit Pith: In Planta Transcriptome and Anatomical Analyses. Front. Microbiol. 2021, 12, 681251. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 September 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.; Liu, Z.A.; He, J.; Jiang, X.; He, F.; Lu, Z.; Yang, S.; Chen, P.; Yu, H.; et al. Somatic Variations Led to the Selection of Acidic and Acidless Orange Cultivars. Nat. Plants 2021, 7, 954–965. [Google Scholar] [CrossRef]
- Wu, G.A.; Prochnik, S.; Jenkins, J.; Salse, J.; Hellsten, U.; Murat, F.; Perrier, X.; Ruiz, M.; Scalabrin, S.; Terol, J.; et al. Sequencing of Diverse Mandarin, Pummelo and Orange Genomes Reveals Complex History of Admixture during Citrus Domestication. Nat. Biotechnol. 2014, 32, 656–662. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The SVA Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
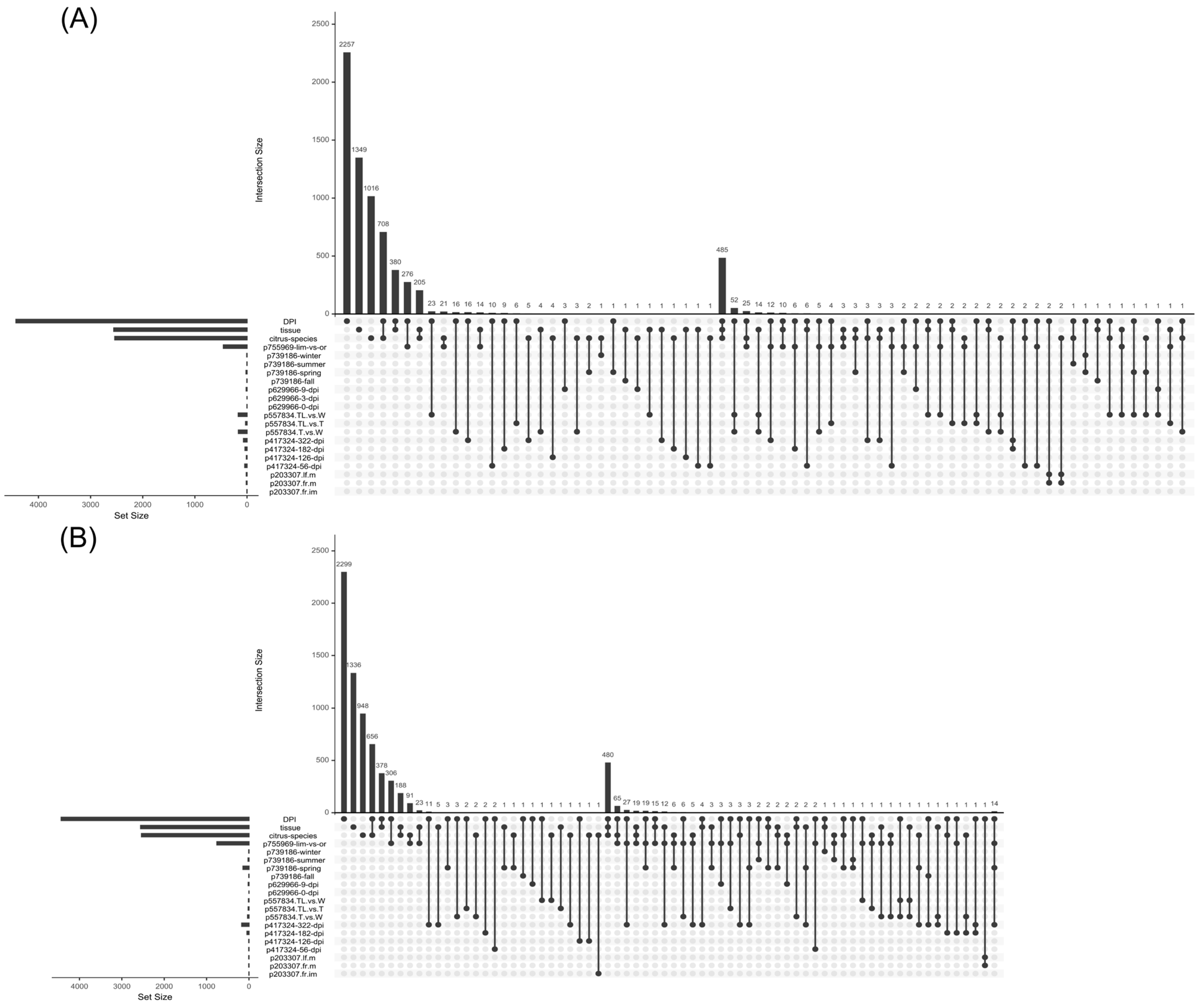
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aligner | Genome | Minimum (%) | Q1 (%) | Median (%) | Q3 (%) | Maximum (%) |
|---|---|---|---|---|---|---|
| STAR | C. sinensis DHSO v3.0 | 23.02 | 63.53 | 88.88 | 92.1 | 95.71 |
| STAR | C. clementina v1.0 | 27 | 66.23 | 89.75 | 92.41 | 95.44 |
| HISAT2 | C. sinensis DHSO v3.0 | 19.17 | 56.05 | 78.06 | 83.3 | 88.6 |
| HISAT2 | C. clementina v1.0 | 14.73 | 55.63 | 80.87 | 85.52 | 91.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, R.; Moschen, S.; Conti, G.; González, S.A.; Rivarola, M.; Gómez, C.; Hopp, H.E.; Fernández, P. Exploring the Genetic Networks of HLB Tolerance in Citrus: Insights Across Species and Tissues. Plants 2025, 14, 1792. https://doi.org/10.3390/plants14121792
Machado R, Moschen S, Conti G, González SA, Rivarola M, Gómez C, Hopp HE, Fernández P. Exploring the Genetic Networks of HLB Tolerance in Citrus: Insights Across Species and Tissues. Plants. 2025; 14(12):1792. https://doi.org/10.3390/plants14121792
Chicago/Turabian StyleMachado, Rodrigo, Sebastián Moschen, Gabriela Conti, Sergio A. González, Máximo Rivarola, Claudio Gómez, Horacio Esteban Hopp, and Paula Fernández. 2025. "Exploring the Genetic Networks of HLB Tolerance in Citrus: Insights Across Species and Tissues" Plants 14, no. 12: 1792. https://doi.org/10.3390/plants14121792
APA StyleMachado, R., Moschen, S., Conti, G., González, S. A., Rivarola, M., Gómez, C., Hopp, H. E., & Fernández, P. (2025). Exploring the Genetic Networks of HLB Tolerance in Citrus: Insights Across Species and Tissues. Plants, 14(12), 1792. https://doi.org/10.3390/plants14121792